Mitochondrial medicine: Fact or fiction? Part II – Mitochondria and the cellular crosstalk.
In this post, we will discuss mitochondrial diseases in more detail. While doing research on this subject I recognized that there are multiple views on how a mitochondrial disease can be defined. Some researchers see mitochondrial diseases or dysfunctions as a result of mutations in genes that are responsible for exact mitochondrial function. Others (like me) assess this in a broader context and would define mitochondrial diseases as all influences suppressing the general cellular function and therefore also the mitochondrial performance.
Hereditary-caused mitochondrial dysfunctions are leading to tremendous problems for the affected persons [1, 2]. This means, most people with mitochondrial dysfunctions are either not able to live (often they die early in their development or childhood) or they are suffering severe health issues throughout their lives. Examples for such problems are deafness, blindness, heart problems, diabetes, liver damages, muscle weakness, fatigue and neurological/psychological problems such as depression or dementia [1-6].
Because of the fact that many of these health issues are multifactorial, it is hard to estimate where the exact cause of the problems is located. However, detailed knowledge of what causes a disease is fundamental for therapy. In fact, mitochondria are prone to multifactorial problems because they are involved in almost every cellular reaction (see the first chapter again).
Mitochondrial dysfunctions, therefore, are an enormous challenge for doctors, scientists and ultimately patients.
Current estimations come to the conclusion that about one in 2000 persons suffer from a mitochondrial dysfunction [1]. Nonetheless, these numbers solely taking people into account which have a mutation in at least one of about 1300 genes that are necessary for mitochondria function (only 37 are encoded in the mitochondria, the remaining genes were outsourced in the nucleus) [1, 2]. However, problems in the cellular crosstalk which also hampers mitochondrial function are not considered [6]. These are, for instance, problems in cellular pathways, recycling mechanisms or the exposure to toxins (antibiotics, heavy metals…) [7].
As a result of the current literature, it is probably impossible to cure mitochondrial dysfunctions, unless you are able to do gene therapies [1, 8]. Even though these kinds of therapies were tested in the past, today they are still far from standard therapy [8]. In addition, the more genes are affected the harder it gets.
Nevertheless, maybe we should get an overview first and then we could probably assess whether any possibilities are present for preventing or curing mitochondrial dysfunctions.
Based on the current literature you can define at least five main causes for mitochondrial dysfunctions:
- Mutations in the mitochondrial DNA related to the origin of certain ethnic groups (present since hundreds or thousands of years).
- Mutations in the mitochondrial DNA introduced more recently.
- Mutations in the mitochondrial DNA accumulated throughout life.
- Mutations in genomic DNA encoding for mitochondria-related gene products.
- Mutations in genes that have an impact on the crosstalk between mitochondria and nucleus.
6. Disturbances within the crosstalk between mitochondria and other organelles, such as the endoplasmic reticulum (ER).
7. Disturbances in metabolism and signaling pathways necessary for the exact mitochondrial function.
I would suggest that we discuss all the problems in detail to see which chances exist to counteract mitochondrial dysfunctions.
Let’s start with the point 1.: Mutations typically for ethnic groups.
These mutations were imprinted in the mitochondrial genome of a defined group of people within many generations. For these people, the mutations are no tremendous disadvantage. Rather these mutations are a benefit to cope with the conditions in their region of origin, the foods they usually consume or other factors typical for various ethnic groups [2]. Accumulations of distinct genetic features in demographic/ethnic groups related to a mutual ancestor are known as haplotypes [2, 4, 9].
Fig.1 Haplotypes of this earth. Note that there are tons of different haplotypes. The picture should simply lighten up the text a bit. If you want to know more about your potential "haplotype membership", just drop in here. Made by Chapper - unrestricted use allowed.
In the past, people's environment had a huge impact on their lives. But in times of sufficient nutrient supply, good infrastructure, excellent hygienic and medical conditions these variations within the mitochondrial genome are not that important anymore [2]. Rather it can cause some trouble such as high caloric foods which are the main cause of obesity and even cancer for some people [4]. Further, longevity is related to certain haplotypes [2].
Whoever wants to know more about their haplotypes can visit the website Ancient mtDNA database or let their mitochondrial genome be sequenced.
I will also do this soon!
To treat these kinds of mutations is probably impossible because of natural selection. All mitochondria within your body bearing these haplotypes. You received them from your mother (mitochondria were transmitted by your mother). Even though there are some doubts about this fact [10].
Same holds true for point 2.: Recently adopted mitochondrial mutations.
According to the literature, you only can cure these kinds of diseases by using genetic tools [1]. The category of recently adopted mitochondrial mutations encompasses, in my opinion, the most severe kinds of mitochondrial dysfunctions. The more mild problems are diabetes type II, low performance while doing sports or deafness [2]. The remaining examples often have tremendous consequences to the affected peoples, such as in the case of MELAS. MELAS is a disease which is characterized by Mitochondria-associated brain damage (Encephalopathies), increased acid in the blood (Lactic acidosis), as well as stroke-like episodes (Stroke). Responsible for these symptoms are often single mutation, for instance in mitochondrial tRNA genes. Fig.2 shows one of the affected tRNAs which carries leucine to the mitochondrial ribosomes (Fig.2) [1, 2].
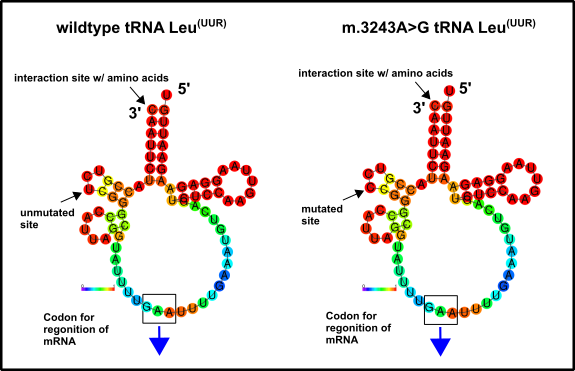
Fig.2 Mitochondrial tRNA which carries the amino acid leucine to the mitochondrial ribosomes. (A) shows the wild type variant and (B) the variant which can be mutated in the disease MELAS. You can find the original mtDNA sequence which encodes this tRNA here. Thereafter, I created a complementary sequence because the process of transcription always produces antiparallel products. This can be carried out with tools like this one here. Subsequently, you have to rewrite the sequence back into an RNA sequence, because RNA carries other nucleotides (e.g. uracil U instead of thymine T). Use the tool right here to realize this. Finally, with the RNAFold WebServer possible models can be illustrated. Shown are binding tendencies or the entropy distribution, for instance. Intriguingly, you can see that there is no severe difference between them. This would implicate that either steric interactions of the tRNA with the ribosome are disturbed by this mutation or that the tRNA folding is disordered. I don’t know but will keep you informed when I have found out more. Made by Chapper - unrestricted use allowed
Everyone should know that leucine is an important amino acid. The absence of this amino acid, therefore, prevents the correct synthesis of mitochondrial proteins [11, 12]. In fact, little mutations such as them have severe consequences, but the curation of problems like this should be no problem in the future (at least in my opinion). You can find similar aspects in the case of the disease MEERF, with even more symptoms but sometimes also only one mutation in genes for mitochondrial tRNAs [1]. A raw assessment reveals that mutations within mitochondrial genes for tRNAs are the primary cause of mitochondrial dysfunctions of category 2. Many affected carriers suffer already in childhood, but the symptoms getting worse with age (see also Charcot–Marie–Tooth disease) [1]. Whoever met people with diseases like this know how hard it is to get a least a bit of life quality. Unbelievable that only one mutated base could be responsible.
Nevertheless, I always ask myself whether the assumption that solely genetic interventions would help is correct. The fact that it is getting worse with age would implicate that there must be also some healthy mitochondria left. Maybe you remember my article about LHON. LHON symptoms (primarily blindness) getting worse with age and some carriers don’t even show symptoms but compensate with increased mitochondrial mass [3]. Could this be a hint for therapy besides genetics?
Ok, let’s move over to category 3.: Mitochondrial diseases you receive during your life. The so-called somatic mutations.
This is a very important category, I think. Let's illustrate this my typical Chappertyle. Here on this blog (WorldofChapper) the reader probably is not familiar with the Packman symbol. But my Steemit followers know what I mean. It's about autophagy. I will add my old autophagy articles also on WorldofChapper, but this will take a while. Therefore just recognize that autophagy is a mechanism within your cells that clears all the junk and waste, including old or malfunctioning mitochondria. The lower your autophagic potential is the more prone you are to diseases such as mitochondrial dysfunctions. Let me briefly illustrate it.
These are your mitochondria when you are young and "healthy":
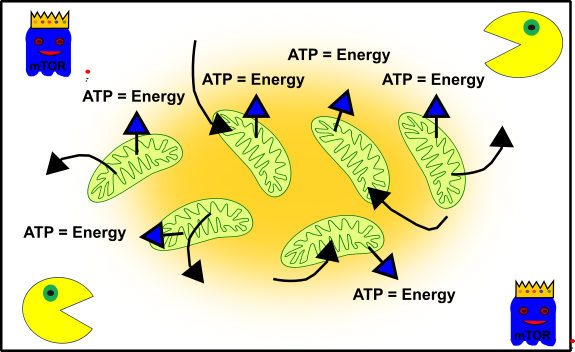
Figure 3: Simplified representation of your cellular structure in young cells. Your mitochondria (green), autophagy (Packman) and cell cycle promoting pathways (ghost in blue with crown). Made by Chapper - unrestricted use allowed
Your mitochondria are busy and diligent. Therefore, you can run around all the time without getting tired. mTOR (the blue ghost) promotes the growth and development of your body. And your cellular packman (autophagy) takes care that everything stays clean and ordered. The perfect symbiosis.
But now you are getting older and everything becomes a bit messy. For instance, you have more grief, eat at McDonald's, start smoking and drinking, or other worse things. The entire cellular network is not that pretty anymore.
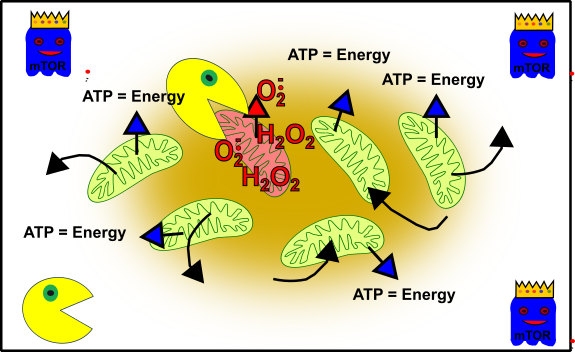
Figure 4: Simplified representation of your cellular structure during your adolescence. Your mitochondria (green), oxidative stress-damaged mitochondria (reddish), autophagy (packman) and cell cycle promoting pathways (ghost in blue with crown). Made by Chapper - unrestricted use allowed
The elevated stress within your cells led to a lot of damage. Oxidative stress (such as by reactive oxygen species, ROS) has knocked these mitochondria out. But don’t panic! Your cellular packman (autophagy) will take care of and enable your cells to stay intact. Everybody knows that life is getting more difficult the older you are and also your cells are more and more challenged throughout the years. Not only your cellular packman becomes lazier [13], also mTOR shows it’s true nature and prevents the death of cells which are completely out of order [14]. Additionally, mTOR works against the cellular packman and therefore worsens the situation [15]:
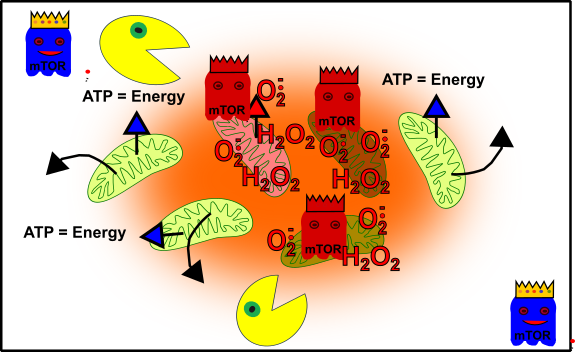
Figure 5: Simplified representation of your cellular structure when you are an adult. Your mitochondria (green), oxidative stress-damaged mitochondria (reddish), autophagy (packman), promoting pathways (ghost in blue with crown), and cellular out of control (red ghosts with crown). Made by Chapper - unrestricted use allowed
Now, you are in trouble: Weakness! Burnout! Everything is difficult! Your performance is bad! No power, nowhere! Then you see all these promising offers about performance-enhancing pills or drugs. Or you think: „Let’s have a little drink, or some candies or junk food to feel better!“. But the truth is that all these things silently killing your cellular integrity. The reason: mTOR is promoted by stress, drugs, bad food (etc.) and this, in turn, suppresses the packman (autophagy). A cascade to death is unleashed! Your mitochondria are on 100% all the time and receiving a lot of damage by the ROS they produce. Unfortunately, no packman recycles them, because he is oppressed by mTOR. In the end, these damaged mitochondria replicating themselves [4]. Eventually, you have some mitochondria in your cells, normally serious ill people have.
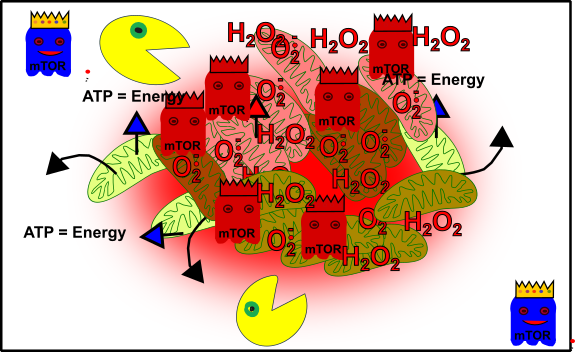
Figure 6: Simplified view: Your cellular structure and health are going to crash! Mitochondria (green), oxidative stress-damaged mitochondria (reddish), autophagy (packman), promoting pathways (ghost in blue with crown) and unbalanced promoting pathways (red ghosts with crown). Made by Chapper - unrestricted use allowed
Congratulations, you successfully ruined your health!
And now? What’s to do? I won’t describe this in detail here. Don’t forget I already create numerous posts concerning this. But soon I will re-issue most of them, so stay with me.
Now to category 4.: Mutations within your nuclear DNA.
This is a very exciting chapter, I think. As I already mentioned in the first article of this series, the mitochondria have outsourced numerous genes in the nucleus of the cell after they arrived in our cells billions of years ago (endosymbiosis) [1, 2, 4, 16]. More than 1300 genes were transferred from the mitochondria to the nucleus. There are only 37 genes left in mitochondria of today [2]. For fully functioning mitochondria you need genes from both origins. In general, the complexes of the respiratory chain are encoded in the nucleus and the mitochondria. But there are also some differences: Complex II, for instance, is encoded solely in the nucleus [2]. No wonder, that problems in the nucleus can also cause trouble.
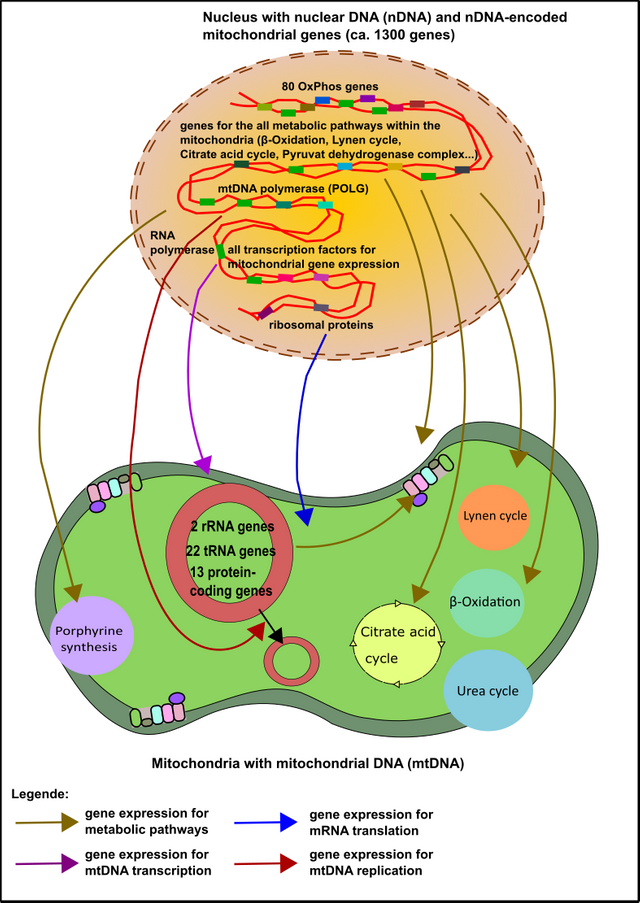
Figure 7: The majority of mitochondrial genes are encoded within the nucleus. These "nuclear genes" are involved in all functions of the mitochondria (enzymatic steps, DNA replication, transcription, translation, ...). Made by Chapper - unrestricted use allowed
The most convincing advantage of the nucleus is the low affinity of the nuclear genes to cellular stress and thus mutations. The exact reason, however, is not completely known. Of course, the close proximity of the mitochondrial DNA to ROS could be one reason (remember that the respiratory chain is the main source of ROS within the cell) [17-19]. Moreover, the mitochondrial genome (mtDNA) is very tiny and completely packed with genes. When some ROS come across it’s no big deal for them to induce DNA damage. Please take into consideration that in contrast to the mitochondrial genome, the nuclear genome carries about 98% non-coding DNA [20]. Many of these sequences are often referred to as „junk“ [20, 21].
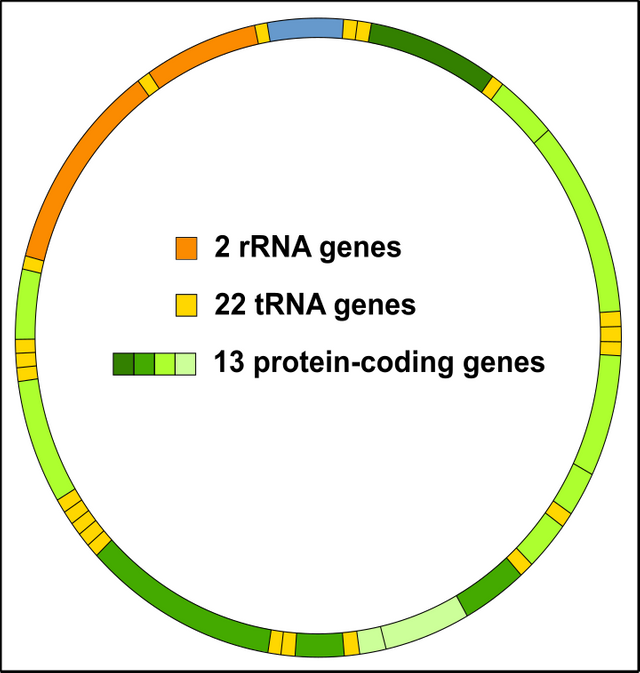
Figure 8: The mitochondrial genome (mtDNA) is densely packed with genes. Every shot is a hit! license-free CC BY-SA 3.0 modified by Chapper.
In essence, you have less stress by ROS per nuclear gene (mtDNA about 1600 to 1700 base pairs vs. nuclear DNA about 2,000,000,000 base pairs) comparable to a "needle in a haystack" [1, 2, 9, 12, 20, 21]. Incidentally, Wallace 2010 [2] has the opinion that the cell does not care about mtDNA, because in nature malfunctioning mitochondria has no chance to survive. Therefore, the cell is not interested in the energy-consuming repairing of mtDNA genes. The latter is far too expensive and the evolution is not primed on the next mTOR boost at McDonald's and Burger King. Therefore, malfunction mitochondria underlie a huge evolutionary pressure. However, the cell probably is not prepared for modern western society.
But I digress because there are certain mutations in mitochondrial genes encodes in the nucleus. The spectrum ranges from serious illnesses such as the Leigh syndrome, which is often fatal in childhood, to depression [1, 2]. Again, no effective therapeutic intervention exists today.
So, let's move on to category 5. and get to the next level: the crosstalk between mitochondria and nucleus.
As you know, the nucleus and mitochondria are in constant exchange with each other. This not only a result of the fact that the mitochondria once exported most of their genetic information to the nucleus [16]. Rather, it is an inevitable fact because mitochondria affect cellular activity, synthesis of components, energy supply, regulation of the ion concentration, as well as life and death [11, 12]. Crosstalk between the nucleus and mitochondria is therefore absolutely crucial because otherwise, the entire cellular structure wouldn't work.
But how can they chat with each other?
At first, of course, by making an agreement to produce more or fewer mitochondria.
The transcriptional co-activator PGC1α is the key to realize this [22]. The reader may remember that transcription factors are those proteins that assist the RNA polymerase specifically search and read genes (please check out here!). Co-activators are those factors which help the transcription factors to better assist the RNA polymerase [23]. In any case, PGC1α is activated under conditions such as cold or hunger, but also after exercise or in the course of oxidative stress [22]. PGC1α then migrates into the nucleus, allowing the synthesis of genes that counteract these "problems". Hunger, for instance, can be compensated by the increased utilization of fatty acids. However, as already mentioned, this is only possible with intact mitochondria (the breakdown of fatty acids is also carried out in peroxisomes but ATP is finally created within the mitochondria) [11, 12]. If the cell freezes, you can uncouple the respiratory chain by using the so-called Uncoupling Protein (UCP). This strategy enables the proton gradient of the mitochondria to produce heat instead of ATP [24]. Oxidative stress comes mainly from damaged mitochondria, therefore you need to produce new. PGC1α carries this out. Moreover, PGC1α provides the crucial impetus for the formation of many key enzymes that counteract oxidative stress (just remember the glutathione system, here) [22]. Thus, the nucleus provides the first basis for fruitful cooperation (Fig. 9).
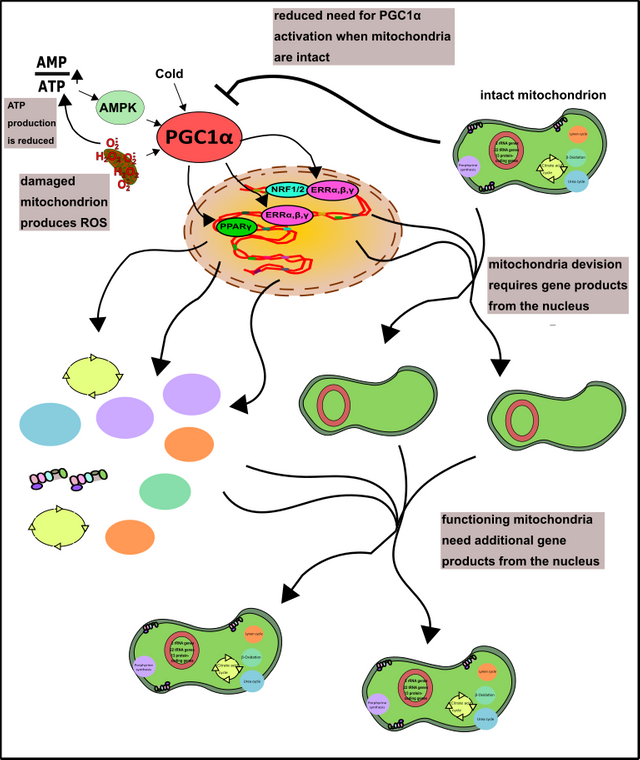
Figure 9: The role of PGC1α in the formation of mitochondria. The often stress-related activity of PGC1α can be diminished by well-functioning mitochondria. More about AMPK you can find here. Made by Chapper - unrestricted use allowed
Furthermore, the nucleus provides additional products and pathways that work around the mitochondria, making their operation possible. These are, for example, the cytoskeleton in which the mitochondria are embedded [25] or the products of glycolysis, amino and fatty acids. All these products not only serve to generate energy (which ultimately benefits the nucleus), these products are of course also precursors of components of the mitochondria themselves (proteins, membranes, etc.) [11, 12, 26, 27].
The mitochondria, in turn, are regulating the function of genes within the nucleus via their metabolic intermediates.
The effect of these products is carried out on an epigenetic level [28]. This means, that not the genetic code but the DNA bases or associated proteins are modified. In turn, these mechanisms enable mitochondria to effectively increase but also decrease the supply of certain gene products from the nucleus. Intermediates from the citric acid cycle such as α-ketoglutarate, fumarate, and succinate, as well as acetyl-CoA, are primarily responsible for epigenetic reprogramming of nuclear genes. Furthermore, even the more frequently mentioned ROS are able to carry out epigenetic modifications and thereby alter the interaction between the nucleus and the mitochondria (Fig.10).
The nucleus, further, can decrease the mitochondrial activity.
For instance, after elevated oxidative stress autophagy can be triggered to remove the ROS-producing mitochondria [4]. Moreover, the nucleus can decrease the synthesis of mitochondria or mitochondrial products for lowering energy production [29].
Additionally, oxygen deficiency is leading to the induction of HIF1 (hypoxia-inducible factor 1) [12]. This happens in your muscles when you exercise [11, 12, 18, 30, 31]. An oxygen deficiency of tissue (hypoxia) is leading to increased ROS production (among others because of the increased degradation of AMP) [18]. Fortunately, there is HIF1, which shuts off the mitochondria and activates glycolysis. This is important because otherwise, the mitochondria would consume the remaining oxygen molecules and thus worsen the situation [32]. The mechanism includes the increased production of glucose transporters (GLUT1) and the increased expression of lactate dehydrogenase. The activation of PDK1 finally switch off the mitochondria. PDK1 is a protein kinase that inhibits pyruvate dehydrogenase in the mitochondria (read more about protein kinases here). As a result, glucose can no longer be metabolized further via the mitochondria (no more aerobic glycolysis is possible, thus no oxygen is wasted). This is one of the reasons why athletes often swear by "fast carbohydrates", as glycolysis (here anaerobic glycolysis) is, as we already know, very inefficient (2 moles of ATP per mole of glucose vs.> 30 moles of ATP) and therefore athletes need it plenty of sugar to meet your needs for ATP. Alternatively, of course, phospho-creatine is an option [31].
It's just important to prevent the accumulation of AMP because it is the origin for further ROS production. Additionally, it takes a few days to produce fresh ADP for ATP production (Fig.10) [31]. Without ADP you can't produce ATP, and AMP can not that easily be used to create ADP (for this you need energy, you don't have).
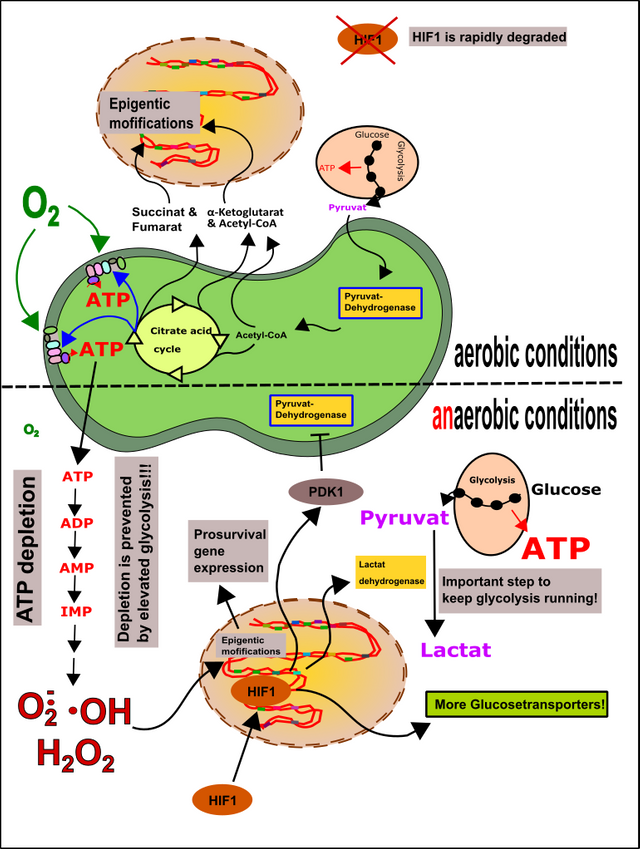
Figure 10: Epigenetics and HIF1. Metabolites of the mitochondria, as well as ROS, can cause epigenetic modifications in the nuclear DNA and thus shape the fate of the cell. Aerobic conditions: HIF1 is rapidly degraded. Anaerobic conditions: HIF1 migrates into the cell nucleus and causes the mitochondria to be switched off while promoting anaerobic glycolysis. The goal is to minimize further oxygen consumption and to generate sufficient amounts of ATP to stop its degradation and thus ROS formation. Made by Chapper - unrestricted use allowed
Worst case: Let the cells die!
In the end, the body should rely on the programmed cell death (apoptosis), e.g. in case of severe DNA damage [12]. This program is carried out by the transcription factor p53. p53 is necessary for the production of Bax. Bax, in turn, generates a channel within the mitochondrial membrane to allow the release of cytochrome c (you remember, it’s one of the molecules which transports electrons in the respiratory chain). Once cytochrome c is released, the so-called apoptosome is built. The apoptosome then destroys all the cellular components, the cell dies (Fig.11).
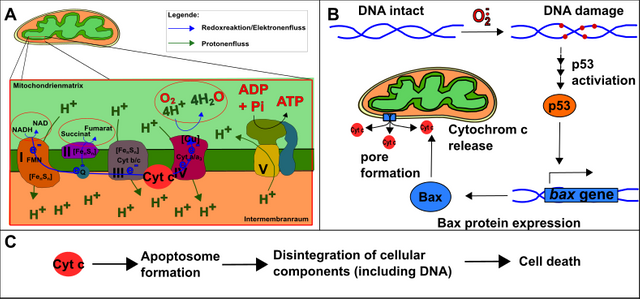
Figure 11: Basis of programmed cell death (apoptosis). A. The respiratory chain with cytochrome c. B. The DNA damage caused e.g. by ROS cause the activation of p53. p53 causes the formation of Bax, which forms a channel in the mitochondrial membrane. This allows cytochrome c to "escape" from the respiratory chain. C. Cytochrome c forms with other factors the apoptosome, which causes cell death. Made by Chapper - unrestricted use allowed
I think this small selection is sufficient (and in fact, there are hundreds, if not thousands, of factors involved) to explain to you that there are many more ways feasible to counteract an optimal function of your mitochondria. Every "brick" in this system is a potential weakness for your mitochondrial performance and, at worst, can lead to diseases such as cancer [2, 4, 12, 33]. Because of these facts, not only the mitochondria itself contribute to phenomena known as „mitochondrial dysfunction“, many other aspects need to be taken into consideration.
Closely linked with the last point is category 6.: Crosstalk of the mitochondria with other organelles.
Actually, this field of research is still very young and less well explored. Therefore, I just want to give some a brief overview of well-investigated findings. Recently, Hirabayashi et al. [34] found that a specific protein called PDZD8 in the membrane of the endoplasmic reticulum (ER) is responsible for the interconnection of the mitochondria with the ER. The ER is not only important for the synthesis of cholesterol and proteins, but it also serves as a calcium reservoir [11, 35]. Calcium, in turn, is a crucial signaling molecule and cofactor of various enzymes. In nerve cells, signals from between are realized via the uptake of calcium. To stabilize a constant calcium concentration in the cytosol, the ER needs to interact with mitochondria. Inbalances in the communication between ER and mitochondria are the basis of problems within the nervous system [35].
But other organelles such as the lysosomes also interact with the mitochondria. Lysosomes are the organelles that are forming our cellular packman and are therefore responsible for autophagy (see here). On the other hand, lysosomes are releasing proteins, such as cathepsins. Cathepsins, in turn, can inhibit the protein Bcl-2, which inhibits Bax [36]. As a result, the lysosomes play a potential role in programmed cell death.
And there are much more mechanisms which also has a huge impact on the function of mitochondria („category 7.“).
You have to understand that all signals generated, all substances released, every ATP molecule formed in turn triggers, promotes, or inhibits other enzymes or reactions. These enzymes once more signal, catalyze or modify other cellular components. In the end, various feedback cycles shaping the cellular landscape. Sounds complicated? Yes, it is, but this network is the basis of all the reactions of your body. I don't want to go too far into detail again, but keep in mind that the noise of millions of molecules determining the balance within your body. This highly regulated concert is meticulously adjusted and therefore also highly susceptible. No wonder that imbalances of this system can disrupt the mitochondrial reaction and thus the entire continuum.
Because of these facts „category 7.“ would be an own chapter. Maybe even more than one article. But I will stop right here, the entire post is far too long anyway.
In conclusion, it would be a much better idea to substitute the term „mitochondrial dysfunctions“ by „cellular dysfunctions“.
Of course, the hardcore of these cellular dysfunctions are the mitochondria. Nevertheless, a unilateral focus on mitochondria is incomplete because important other areas are not taken into account.
You are no mitochondria on two legs!
Let’s summarize this:
Mitochondria are the hub of all cellular events (I'm not saying metabolism). The DNA of the mitochondria (mtDNA) is tightly packed with genes that can rapidly mutate through ROS produced by mitochondria itself. Some of these mutations have been conserved as haplotypes in geographically definable populations and are a selective advantage for them. Other mutations are "recent" and may cause severe diseases such as MELAS, MERRF or LHON. Since many diseases get worse over the course of life, the selection for remaining intact mitochondria could be therapeutically useful. At the moment, however, gene therapy should be the intervention of choice in the future. However, "mitochondrial selection" might be a well-working approach to prevent mitochondrial and/or cellular dysfunctions. Keep in mind, that mitophagy of damaged mitochondria (autophagy of mitochondria) is a scientifically proven fact.
And there are even more mechanisms that can be linked to mitochondrial dysfunctions (and should be termed cellular dysfunctions). A very prominent example is a disease called Porphyria variegata. Porphyria variegata (porphyria) is a disorder in porphyrin synthesis, which occurs in part in mitochondria (the last two steps) [33]. Since porphyrins are (among others) important for the transport of oxygen in the red blood cells, such a disease can have severe consequences. The symptoms range from itching, pain, muscle weakness to neurological deficits [37]. It is believed today that both King George III. from England as well as Count Dracula (Vlad III.) suffered from porphyria. Some researchers argue, this explains England's catastrophic diplomacy, which ultimately led to the War of Independence and the founding of the United States. Also, Count Dracula's propensity to impalement could be explained by "temporary" mental problems caused by porphyria [37].

Fig. 12: Dracula (CC0, from Pixabay), mechanism of oxygen binding and flag oft he USA (both designed by Chapper).
You can move on in this manner but the final conclusion is that 1. mitochondria are involved in many cellular settings and 2. a therapy is hard to implement because of the fact that mitochondrial dysfunction can have various origins. Nevertheless, in the next chapter, we will try to find ways for diagnostics and therapy of these diseases.
So far
Take care and
May Da Mitos Be With You.

Chapper
Oh, before I forget:
The title of this series raises the question of whether mitochondrial medicine is a fact or fiction. I just want to argue in this way: While you are reading these lines about 100 g of ATP is in your body. Within one day about 100 kg are produced [31]. After combining these facts with all the facts we collected today it should be clear that mitochondria and mitochondrial health is a scientifically proven fact.
References:
- Suomalainen, A. and B.J. Battersby, Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat Rev Mol Cell Biol, 2018. 19(2): p. 77-92.
- Wallace, D.C., Mitochondrial DNA mutations in disease and aging. Environ Mol Mutagen, 2010. 51(5): p. 440-50.
- Kirches, E., LHON: Mitochondrial Mutations and More. Curr Genomics, 2011. 12(1): p. 44-54.
- Hill, B.G., et al., Integration of cellular bioenergetics with mitochondrial quality control and autophagy. Biol Chem, 2012. 393(12): p. 1485-1512.
- Brand, M.D. and D.G. Nicholls, Assessing mitochondrial dysfunction in cells. Biochem J, 2011. 435(2): p. 297-312.
- Myhill, S., N.E. Booth, and J. McLaren-Howard, Chronic fatigue syndrome and mitochondrial dysfunction. Int J Clin Exp Med, 2009. 2(1): p. 1-16.
- Michalak, K., et al., Treatment of the Fluoroquinolone-Associated Disability: The Pathobiochemical Implications. Oxid Med Cell Longev, 2017. 2017: p. 8023935.
- Lewis, R., Gentherapie, zweiter Anlauf. Spektrum der Wissenschaft Spezial Biologie Medizin Hirnforschung, 2016. 2/2016.
- Ehler, E., et al., AmtDB: a database of ancient human mitochondrial genomes. Nucleic Acids Res, 2019. 47(D1): p. D29-D32.
- McWilliams, T.G. and A. Suomalainen, Mitochondrial DNA can be inherited from fathers, not just mothers. Nature, 2019. 565(7739): p. 296-297.
- Müller-Esterl, W., Biochemie: Eine Einführung für Mediziner und Naturwissenschaftler. 2004: Spektrum Akademischer Verlag.
- Püschel, Taschenlehrbuch Biochemie. 2011: Thieme Verlagsgruppe.
- Metaxakis, A., C. Ploumi, and N. Tavernarakis, Autophagy in Age-Associated Neurodegeneration. Cells, 2018. 7(5).
- Laplante, M. and D.M. Sabatini, mTOR signaling at a glance. J Cell Sci, 2009. 122(Pt 20): p. 3589-94.
- Kast, B., Der Ernährungskompass: Das Fazit aller wissenschaftlichen Studien zum Thema Ernährung - Mit den 12 wichtigsten Regeln der gesunden Ernährung. 2018: C. Bertelsmann Verlag.
- Embley, T.M. and W. Martin, Eukaryotic evolution, changes and challenges. Nature, 2006. 440(7084): p. 623-30.
- Mari, M., et al., Redox control of liver function in health and disease. Antioxid Redox Signal, 2010. 12(11): p. 1295-331.
- Kalyanaraman, B., Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol, 2013. 1(1): p. 244-57.
- Naviaux, R.K., Metabolic features of the cell danger response. Mitochondrion, 2014. 16: p. 7-17.
- Palazzo, A.F. and T.R. Gregory, The case for junk DNA. PLoS Genet, 2014. 10(5): p. e1004351.
- W. Janning, E.K., Genetik: Allgemeine Genetik - Molekulare Genetik - Entwicklungsgenetik. 2004: Georg Thieme Verlag.
- Austin, S. and J. St-Pierre, PGC1alpha and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci, 2012. 125(Pt 21): p. 4963-71.
- Lin, J., C. Handschin, and B.M. Spiegelman, Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab, 2005. 1(6): p. 361-70.
- Chen, W., Q. Yang, and R.G. Roeder, Dynamic interactions and cooperative functions of PGC-1alpha and MED1 in TRalpha-mediated activation of the brown-fat-specific UCP-1 gene. Mol Cell, 2009. 35(6): p. 755-68.
- Kuznetsov, A.V., et al., Cytoskeleton and regulation of mitochondrial function: the role of beta-tubulin II. Front Physiol, 2013. 4: p. 82.
- Mileykovskaya, E. and W. Dowhan, Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta, 2009. 1788(10): p. 2084-91.
- Horvath, S.E. and G. Daum, Lipids of mitochondria. Prog Lipid Res, 2013. 52(4): p. 590-614.
- Matilainen, O., P.M. Quiros, and J. Auwerx, Mitochondria and Epigenetics - Crosstalk in Homeostasis and Stress. Trends Cell Biol, 2017. 27(6): p. 453-463.
- Cagin, U. and J.A. Enriquez, The complex crosstalk between mitochondria and the nucleus: What goes in between? Int J Biochem Cell Biol, 2015. 63: p. 10-5.
- al., G.e., Taschenlehrbuch Physiologie. 2. Auflage ed. 2015: Thieme.
- Booth, N.E., S. Myhill, and J. McLaren-Howard, Mitochondrial dysfunction and the pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). Int J Clin Exp Med, 2012. 5(3): p. 208-20.
- Okamoto, A., et al., HIF-1-mediated suppression of mitochondria electron transport chain function confers resistance to lidocaine-induced cell death. Sci Rep, 2017. 7(1): p. 3816.
- Peter Karlson, D.D., Jan Koolman, Georg Fuchs, Wolfgang Gerok, Ruth Hammelehle, Karlsons Biochemie und Pathobiochemie. Vol. Auflage: 15. 2005: Thieme.
- Hirabayashi, Y., et al., ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science, 2017. 358(6363): p. 623-630.
- Lombardi, A.A. and J.W. Elrod, Mediating ER-mitochondrial cross-talk. Science, 2017. 358(6363): p. 591-592.
- Repnik, U. and B. Turk, Lysosomal-mitochondrial cross-talk during cell death. Mitochondrion, 2010. 10(6): p. 662-9.
- Hill, F.E.D.E.D., Porphyrine: Ein Ring für die Farben des Lebens. Spektrum der Wissenschaft Biologie Medizin Hirnforschung, 2019. 1/19.
Posted from my blog with SteemPress : http://worldofchapper.de/rp295320-ovh/index.php/2019/08/16/mitochondrial-medicine-fact-or-fiction-part-ii-mitochondria-and-the-cellular-crosstalk/
Chapper is back!! Haven't read the blog. Just glad to see you here. Looking forward to this. English--it should be a breeze. I'll edit this comment later with meaningful feedback after I've taken my time to digest what I know will be valuable information.
Hope your summer is going well.
Warm regards,
AG
Hey @agmoore, yes I finally realized the creation of a new article. But I have to admit that while making this post I recognized that my articles are brutally long. It is actually more torture for readers and storage of information than relaxed knowledge transfer.
Anyway, I did it.
Have a nice weekend
Chapper
Such a mischaracterization. You're a bridge that connects the conversational with the technical. Between you and @scienceblocks, I'm getting the equivalent of an introductory course in cell biology. And it's painless. How can there be too much information? The articles are not full of jargon. After reading yours I looked up lysosome and found an article that was difficult to understand, that depended on technical terminology to transmit information. And that article did not have colorful illustrations to soften the impact :)
When a scientist writes for SteemStem, the challenge is to write for people like me (non scientists) and yet offer material that will be informative to the science community. A delicate balance, and in my view you meet it every time.
It really is a pleasure to find your article here. Thank you for taking precious time to share what you know and explain this information in a way that is accessible.
Later I go to @chapzilla to see if I can sort the German out :)
Have a wonderful day. I'm looking forward to the next chapter.
Regards and Respect,
AG
Hey @agmoore2,
thanks again! Really appreciate your feedback, it's really impressive that you take the time to study all these aspects in detail, respect. The next chapter will take some time. I also have plans to re-issue some of my old articles (you probably haven't read them, we were not connected at this time). Most of them are pretty long and full of information. In the future, I plan shorter articles not just because of the readers, also because of me ;-)
A wonderful weekend also to you
Regards
Chapper
🌟
I really enjoy reading those posts… and actually what I enjoyed the most are the home-made pictures (especially the pacman ones). :)
I have a small question this time:
This corresponds to a huge fraction of the population, doesn’t it? But I guess those numbers account both from the positive mutations and negative ones all together (cf. your point 1).
Hey @lemouth,
it's fine that you are enjoying my pix.
Concerning your question:
No, the number corresponds primarily to category 2. & 4.
Everyone belongs to category 1., that's your heritage and in general, you don't suffer from them.
Nevertheless, it is a variation in function and could, therefore, be a potential problem.
But you are right 1 in 2000 are a lot and there are much more people affected because this is a number belongs to clear characterized mutations. Other problems are not taken into account thus far!
Regards,
Chapper
Ok sorry, I got it wrong. But do we agree that this 1/2000 always refers to dysfunctions. As a mutation does not necessarily imply a dysfunction, doesn't it?
You got it!
This post has been voted on by the SteemSTEM curation team and voting trail.
If you appreciate the work we are doing, then consider supporting our witness stem.witness!
For additional information please join us on the SteemSTEM discord and to get to know the rest of the community!
thanks guys :-)
Thanks for information about medicine.
Hi, @chappertron!
You just got a 0.22% upvote from SteemPlus!
To get higher upvotes, earn more SteemPlus Points (SPP). On your Steemit wallet, check your SPP balance and click on "How to earn SPP?" to find out all the ways to earn.
If you're not using SteemPlus yet, please check our last posts in here to see the many ways in which SteemPlus can improve your Steem experience on Steemit and Busy.
@chappertron You have received a 100% upvote from @steemconductor because this post did not use any bidbots and you have not used bidbots in the last 30 days!
Upvoting this comment will help keep this service running.
Congratulations @chappertron! You have completed the following achievement on the Steem blockchain and have been rewarded with new badge(s) :
You can view your badges on your Steem Board and compare to others on the Steem Ranking
If you no longer want to receive notifications, reply to this comment with the word
STOP