BCL-2 homology and Signal transduction in relation to mitochondrial-dependent apoptosis, and neoplasia (part 6)
6.0 P53, guardian of the genome and potent regulator of apoptosis
There are a lot of important proteins involved in the regulation of apoptosis outside of the BCL-2 family. The tumour suppressor protein p53 is a key regulator of DNA damage-induced apoptosis and works in synergy with many members of the BCL-2 family to induce apoptosis. Upon induction of DNA damage, cellular stress or hypoxia p53 functions by activating the DNA repair proteins and causing cell cycle arrest. Cell cycle arrest is caused by p53 trancriptionally promoting p21 which binds to and inhibits cyclin dependent kinases (CDK), such as CDK2 preventing the cell from entering the synthesis phase. If DNA damage is irreparable p53 can initiate apoptosis through multiple pathways. The molecular mechanism by which p53 induces apoptosis remains to be fully elucidated.
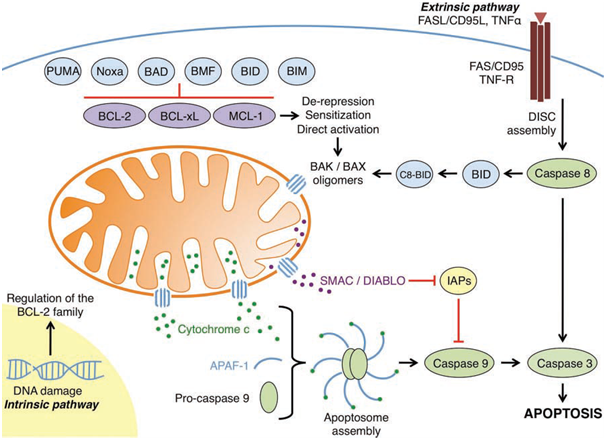
Figure 1: An overview of the intrinsic and extrinsic apoptotic pathways
(Elkholi et al., 2011).
In unstimulated cells p53 levels are kept low by ubiquitination and degradation. A transcriptional target of p53 is the protein murine double minute 2 (Mdm2). MDM2 marks p53 for degradation by the proteasome. This negative feedback loop keeps p53 levels low and prevents transcription of pro-apoptotic proteins (Wee et al 2009).
Interaction between p53 and Mdm2 involve three crucial hydrophobic residues of p53 (phenylalanine-19, tryptophan-23, and leucine-26). One mechanism of interaction between the BCL-2 family is by binding of residues on the BH3 domain into hydrophobic pockets forming strong van der Waals interaction. Interaction of p53 with the BCL-2 family occurs via the hydrophobic residues of p53 occupying the hydrophobic pockets of BCL-2 members, this occurs in a similar manner to that of the BH3 domain. This interaction has been shown recently between p53 and BCL-xL (Bharatham et al 2011).
The tumour suppressor protein p53 transcriptionally targets a plethora of genes. Noxa and PUMA are transcriptionally expressed by p53 during DNA damage (Oda et al., 2000: Wang et al., 2007). PUMA is critical in the ability of p53 to induce apoptosis. PUMA displaces p53 from the BCL-xL:p53 complex, allowing p53 to induce MOMP by either directly activating BAX or BAK itself or by freeing other sequester direct activators (Bharatham et al., 2011). Another BCL-2 family member that is transcriptionally targeted by p53 is the pro-apoptotic BH3-only protein BAD. BAD induces MOMP by binding with the BCL-2 anti-apoptotic proteins freeing sequestered p53. BAD is not only transcriptionally expressed by p53, but also interacts with cytoplasmic p53 (Jiang et al., 2006). It interacts with p53 in a negative feedback loop similar to that of p53 and Mdm2, although with pro-apoptotic effects. BAD prevents cytoplasmic p53 from translocating to the nucleus, preventing increased transcriptional expression of BAD. BAD directs p53 to the OMM and facilitates MOMP through activation of BAK.
BAX and BID are also transcriptionally expressed by p53, showing the wide range of mechanisms in which p53 can induce apoptosis in a transcriptionally-dependent manner.
Induction of apoptosis by p53 can also occur in a transcriptionally-independent manner. The direct activator BIM has been shown to be required for p53-mediated mitochondrial apoptosis (Han et al., 2010). BIM can be sequestered by anti-apoptotic proteins such as MCL-1, BCL-2, and BCL-xL; p53 can function as a BIM derepressor, releasing BIM from sequestration. This allows BIM to directly activate BAX and/or BAK resulting in MOMP. BAX and BAK can also be directly activated by p53, showing that p53 can act as a direct activator or a derepressor. Whether it predominantly acts as a direct activator or depressor is not known, and perhaps it exerts it transcriptionally-independent pro-apoptotic effect by functioning in both ways. Or perchance the function of p53 in this regard is dependent on the cell type or the stimuli that has induced a response.
Another protein that is crucial to regulation of apoptosis is Akt. Whereas p53 is a pro-apoptotic protein, Akt is an anti-apoptotic protein. Akt targets many proteins linked to apoptosis such as HRK and Apaf1. A common target for both p53 and Akt is BAD. Akt inhibits BAD via phosphorylation, preventing it from interacting with anti-apoptotic BCL-2 family members and from directing p53 to the OMM. This convergence of a common target for both Akt and p53 highlights the importance of BAD at inducing apoptosis. High levels of p53 and low levels of Akt predict a cell in a pro-apoptotic state, whereas low levels of p53 and high levels of Akt predict a pro-survival state (Wee et al., 2009).
Another transcriptional target of p53 is the anti-apoptotic protein, apoptosis repressor with caspase recruitment domain (ARC). ARC can block the intrinsic and extrinsic death pathway and is transcriptionally repressed by p53 (Li et al., 2008). As mentioned previously PUMA and BAD are upregulated by p53. The pro-apoptotic function of PUMA and BAD can be inhibited by ARC which interacts with the N-terminal of these BH3-only proteins. This shows that p53 can initiate apoptosis by transcriptionally suppressing the expression of the anti-apoptotic protein ARC while simultaneously upregulating pro-apoptotic proteins such as BAD and PUMA. The downregulation of ARC prevents ARC-induced inhibition of BAD and PUMA. This highlights one of the many pathways in which p53 induces apoptosis.
Part 1
Part 1.1-2
part 2
part 3
part 4
part 5
part 7
@RiskDebonair
Adventure Capitalist of the Future
This post has been ranked within the top 80 most undervalued posts in the second half of May 21. We estimate that this post is undervalued by $13.57 as compared to a scenario in which every voter had an equal say.
See the full rankings and details in The Daily Tribune: May 21 - Part II. You can also read about some of our methodology, data analysis and technical details in our initial post.
If you are the author and would prefer not to receive these comments, simply reply "Stop" to this comment.