Xeroderma pigmentoso | Caso clínico con fotos originales
Hoy les compartiré un caso clínico que realice el año pasado, este fue hecho en base a una niña que asistió al hospital donde realizo mis practicas médicas.
Xeroderma pigmentoso
Introducción
El xeroderma pigmentoso (XP) es una enfermedad autosómica recesiva infrecuente que se origina por un defecto en el sistema REN (reparación por escisión nucleotídica). Este es el sistema reparativo más importante para preservar al DNA de los daños por factores ambientales, químicos y físicos, cuya característica principal es que involucra el recorte, no solo de las bases dañadas del DNA, sino de un trecho bastante largo de alrededor de 30 nucleótidos de la cadena del DNA que comprende las bases dañadas y su región aledaña.
El XP es provocado por una excesiva sensibilidad a los efectos de las radiaciones ultravioleta, esto conlleva a la formación de dímeros de pirimidina en el DNA de las células de la piel. El sistema REN en individuos normales suele reparar estos dímeros; en cambio, en los pacientes afectados con xeroderma pigmentosa el sistema enzimático no funciona adecuadamente.
Dentro de las enzimas reparadoras implicadas en la corrección de los dímeros de timina tenemos las helicasas, que desenrollan la doble hélice de DNA, una endonucleasa que corta el DNA en el sitio del dímero y los nucleótidos adyacentes, una polimerasa que rellena el hueco con las bases de DNA y una ligasa que une de nuevo la porción corregida del DNA con la cadena original. Una mutación heredada en cualquiera de los 7 genes que codifican las enzimas reparadoras puede producir xerodermia pigmentosa.
La incidencia mundial de la enfermedad es de 2 a 4 nacidos vivos por millón. El XP se clasifica en 8 grupos complementarios (A, B, C, D, E, F, G y V [variable]) de acuerdo a las manifestaciones clínicas y a las alteraciones genéticas involucradas, donde el grupo A es el más severo mientras que el grupo C es el más frecuente.
Dentro de las manifestaciones clínicas más resaltantes se encuentran las alteraciones cutáneas que consisten en eritema, descamación, ampollas, costras, efélides, telangiectasias y queratosis actínica, las cuales suelen presentarse primero durante la etapa del lactante o la primera infancia, en las zonas expuestas al sol, tales como la cara, el cuello, las manos y los brazos; no obstante, pueden aparecer en otros lugares, incluido el cuero cabelludo. Entre las manifestaciones oculares figuran fotofobia, lagrimeo, blefaritis, simbléfaron, queratitis, opacidad corneal, tumores palpebrales y posibilidad de evolución a ceguera. Aproximadamente el 20% de los pacientes presenta anomalías neurológicas, como retraso mental y sordera neurosensorial.
Los pacientes con XP menores de 20 años de edad, tienen un riesgo 1000 veces mayor de presentar carcinoma basocelular, carcinoma espinocelular o melanoma cutáneo. Esta enfermedad es sumamente mutilante y la esperanza de vida se encuentra a menudo, disminuida.
El XP se observa con frecuencia cuando hay consanguinidad, por lo que es importante el diagnóstico prenatal y prevenir las posibles complicaciones que se puedan presentar. Sin embargo, el diagnóstico prenatal de esta afección no se efectúa en el país y sólo un adecuado asesoramiento a los padres puede lograr que ellos adopten una correcta conducta reproductiva.
Caso clínico
Se trata de paciente femenina de 13 años de edad, procedente del Estado Trujillo, con antecedentes de hepatitis A (2 años), raquitismo hipofosfatémico (8 años), hipercalciuria (8 años) y litiasis renal bilateral no obstructiva (12 años), hija de padres consanguíneos (primos segundos), sin antecedentes patológicos familiares; producto de la primera gestación, de embarazo controlado a partir de los dos meses, obtenida por cesárea a las 39 semanas de gestación, con un peso de 2.050 gr y una talla de 49 cm, con llanto al nacer, buena succión y alimentación, no presentó cianosis.
A los 8 días de nacida la madre refiere cambios en la apariencia de la piel en regiones expuestas al sol, por lo cual acude al Hospital “Dr. José Gregorio Hernández”, donde es valorada, presentando al examen físico múltiples lesiones vesiculosas y ampollares. A los 2 meses es hospitalizada con clínica similar, donde se realiza una biopsia de piel reportando dermatosis ampollar Ig lineal crónica de la infancia, por lo que se le prescribe dapsona y prednisona.
Al año de edad presentó descamación y aumento del grosor de los pliegues corneales de la piel del cuello (posterior), xerosis y múltiples efélides agrupadas, hipocrómicas en cara, cuello, tronco y extremidades inferiores; debido a los cambios en las lesiones de la piel, se solicita una nueva biopsia, siendo compatible con xeroderma pigmentosa. Durante su evolución, la paciente mostró un avance progresivo de la enfermedad, con agravamiento de las lesiones de la piel, fotofobia y retardo en el crecimiento.
Actualmente, presenta un peso de 12 kg, una de talla 100 cm y una circunferencia cefálica de 45.5 cm, encontrándose todos estos valores por debajo del percentil 3 (figura 1). Al examen físico presenta raíz nasal elevada, pabellones auriculares bien implantados, sin deformidades, cuello móvil, sin tumoraciones, tórax simétrico, murmullo vesicular audible en ambos campos pulmonares, ruidos cardíacos rítmicos sin auscultación de soplos, abdomen blando, depresible, no doloroso a la palpación, poco desarrollo de tejido muscular y adiposo, genitales femeninos con escaso vello púbico y ausencia de menarquia.
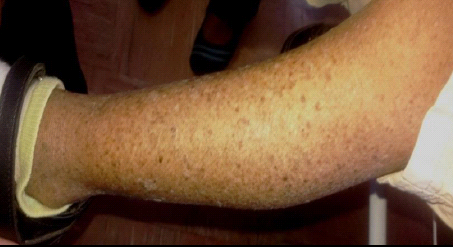
En la inspección dermatológica se observan lesiones múltiples de fotodaño, no dolorosas a la palpación, algunas de ellas hipocrómicas y otras hipercrómicas, en áreas cubiertas y expuestas que se acompañan de queratosis actínica, atrofia y xerosis (figuras 2 y 3). Al examen oftalmológico, presenta hendiduras palpebrales horizontalizadas, fotofobia y queratitis en ambos ojos. A la evaluación neurológica, se evidenció microcefalia, retardo en el desarrollo psicomotor, déficit auditivo e hiporreflexia.
El tratamiento que recibe la paciente está orientado a evitar en lo posible la exposición a los rayos solares. Debe mantener en forma continua fotoprotección e hidratación con cetaphil spf 30 – 50 por tiempo indefinido. Es tratada con systane ultra (1 gota en ambos ojos cada 2 horas) y acrylarm (1 gota en ambos ojos por la noche) de forma permanente, debido a sus manifestaciones oculares y mantiene un tratamiento con citrato de potasio 10.8 % (12cc después del desayuno y almuerzo), sulfato de zinc 0,25% (10 cc después de la merienda y cena) y Rocaltrol cápsula 0,25 mcg, prescrito por el nefrólogo.
La paciente es valorada cada 6 meses en la consulta de genética médica y nefrología infantil del Hospital Universitario “Dr. Pedro Emilio Carrillo”, así como en los servicios de dermatología sanitaria y oftalmología del Hospital “Dr. José Gregorio Hernández”. En los controles médicos llevados a cabo, se ha observado un deterioro en su estado físico y neurológico que ha ido progresando, lo cual indica un mal pronóstico. Además de esto, se ha visto un aumento en las lesiones de la piel con características neoplásicas, específicamente en el 1/3 medio del antebrazo izquierdo, región plantar izquierda y puente nasal, algunas de ellas hiperpigmentadas, hiperqueratósicas, elevadas, de bordes irregulares, pero a las mismas no se les ha hecho el estudio histopatológico respectivo.

Figura 1. Foto actual de la paciente, donde se observa una talla baja para su edad
Figura 2

Figura 3
Se evidencian las lesiones hiperqueratósicas, algunas de ellas hipercrómicas e hipocrómicas con presencia de xerosis en mano derecha y pie izquierdo, respectivamente.
Discusión
El xeroderma pigmentoso tiene una frecuencia en los Estados Unidos de América y Europa de 1:250 000; en Venezuela no hay registros oficiales de esta enfermedad. El caso presentado es el primero en ser reportado en el Hospital Universitario “Dr. Pedro Emilio Carrillo” y por ello, es importante que los médicos tengan en cuenta las manifestaciones clínicas de esta alteración genética, ya que la misma está presente en nuestro estado, así el personal al momento de la consulta estará en la capacidad de detectar y establecer un diagnóstico adecuado en el momento oportuno.
El origen genético del XP se explica porque el copiado del ADN durante las múltiples divisiones celulares, es un proceso propenso a sufrir errores. Son muchos los factores que pueden ocasionar el deterioro del ADN; por ejemplo, la luz ultravioleta puede producir dimerización entre residuos de timina adyacentes en la misma cadena de ADN, de manera que los nucleótidos ya no tienen la capacidad de aparearse con los residuos de adenina de la cadena complementaria y las mutaciones que afectan esta capacidad de reparación son la causa del trastorno genético.
Hay tres etapas evolutivas y morfológicas del XP: en la primera, o fase eritematopigmentaria; hay eritema, edema, y en ocasiones vesículas y ampollas, después aparecen abundantes manchas lenticulares de color café, con tendencia a confluir. En la segunda etapa o fase atrófica-telangiectásica; estas lesiones se acompañan de adelgazamiento de la nariz, mutilación de los pabellones auriculares y microstomía, también hay verrugosidades y queratosis actínicas. En la tercera etapa, o tumoral, sobrevienen epiteliomas basocelulares o espinocelulares, queratoacantomas, sarcomas, melanomas (5%) o diferentes neoplasias benignas.
En la primera etapa, la paciente presentó vesículas y ampollas como lesiones principales, seguidas de múltiples efélides hipocrómicas e hipercrómicas. En la segunda etapa, la característica principal fue la presencia de queratosis actínica. A la paciente se le han extraído lesiones como nevos celulares, pero debido a las condiciones socioeconómicas de los familiares, no se realizaron los estudios histopatológicos correspondientes para el descarte o confirmación de carcinoma basocelular, espinocelular o melanoma maligno, por lo tanto, no se clasifica como en etapa tumoral.
La paciente en sus primeros años de vida tuvo un desarrollo psicomotor normal, caminó a los 9 meses y sus primeras palabras fueron expresadas a los 12 meses. Pero debido a la progresión de la enfermedad, se ha visto un deterioro marcado en su desarrollo, presentando microcefalia y retardo mental de moderado a severo; esto concuerda con lo establecido en la literatura consultada, ya que se han comunicado anomalías neurológicas aproximadamente en el 30% de los pacientes. El inicio suele ser temprano en el primer año de vida, o en algunos pacientes puede diferirse a la segunda década. Las anomalías neurológicas pueden ser leves o graves, con retardo mental progresivo, sordera neurosensorial, espasticidad o convulsiones.
En cuanto a las manifestaciones oculares, en este caso las más importantes han sido fotofobia y queratitis crónica, que han conducido a un déficit de la agudeza visual y son una característica importante del XP. Los hallazgos clínicos se encuentran notablemente limitados a estructuras anteriores, como párpados, córnea y conjuntiva, expuestas a la radiación UV.
El diagnóstico de esta enfermedad es generalmente clínico y se confirma mediante el estudio histopatológico de las lesiones de piel, como se realizó en la paciente, incluso el diagnóstico puede ser en etapa prenatal, midiendo la síntesis de DNA no programada inducida por radiación UV, en células del líquido amniótico cultivadas y con el uso del diagnóstico de DNA de células de trofoblasto obtenidas en etapas tempranas del embarazo, pero estas técnicas sólo son usadas en los países desarrollados. Los diagnósticos diferenciales principales son otras dermatosis inducidas o agravadas por la luz o enfermedades hereditarias con aumento de la hipersensibilidad celular: S. de Cokayne, S. de Bloom, S. de Rothmun-Thompson, Progeria.
El tratamiento está orientado hacia las medidas preventivas, o sea, evitar la exposición a las radiaciones solares, así como los efectos que causan los radicales libres: restricción de iluminación solar mediante el empleo de ropa gruesa o doble, evitar el reflejo solar del agua o de la nieve, el cabello largo, gafas anti-rayos ultravioleta, loción bloqueadora con factor de protección antisolar, climatización del cuarto donde habita el niño para evitar que sude o prolongar así la acción de las lociones protectoras . En la actualidad, no hay un tratamiento curativo para la XP, por lo que se debe informar sobre la enfermedad al paciente y/o encargados familiares o tutores; practicar exámenes médicos periódicos, la extirpación precoz de toda lesión/tumor sospechosos y consejo genético (recordar a los padres que tienen una posibilidad en cuatro de tener otro hijo con la misma enfermedad).
Debido a la situación económica de la familia no se han realizado todos los estudios complementarios que se ameritan y no se han tomado todas las medidas de prevención requeridas. Esto ha llevado a que la evolución del XP en la paciente sea rápidamente progresiva y del mal pronóstico.
Referencias Bibliográficas
1.- Morelli J. Fotosensibilidad. En: Kliegman R, Jenson H, Stanton B, Behrman R. Nelson Tratado de Pediatría. 18° Edición. España: Editorial Elsevier. Año 2013. P: 2335.
2.- Solari A, Roubicek M. Genética Humana. Fundamentos y aplicación en medicina. 4°Edición. Argentina: Editorial Medica Panamericana. Año 2011. P: 165-169.
3.- Jorden L, Carey J, Bamshad M, White R. Genética Médica. 2° Edición. España: Editorial Harcourt. Año 2000. P. 109.
4.- Camargo R, Choque J, Magne W, Madariaga J. Xeroderma pigmentoso. Revista Boliviana de Pediatria. [Internet]. 2008 [citado el 28 de Noviembre 2017]; 47 (1): 16-18. Disponible aquí
5.- Liy M, Duran C, Orozco L, Saez M, Carrasco D, Ruiz M. Xeroderma Pigmentosa con retraso Psicomotor. Reporte de 2 casos de origen Mexicano. Dermatología Pediátrica Latinoamericana. [Internet] 2004, [citado el 28 de Noviembre 2017]; 2 (1) 50 – 53. Disponible aquí
6.- Wolff K, Goldsmith L, Kats S, Gilchrest B, Paller A, Leffell D. Fitzpatrick. Dermatología en Medicina General. 7° Edición. Argentina: Editorial medica panamericana. Año 2009. P. 1315 – 1321.
7.- Diaz D, Herrera A, Vigera M. Xeroderma Pigmentoso. Presentación de un caso. Revista Electrónica de las Ciencias Médicas en Cienfuegos. [Internet] 2008 [citado el 28 de Noviembre 2017]; 6 (2): 64 - 67. Disponible aquí
8.- Falcón L, Dorticós A, Simón R, Garbayo E. Xeroderma Pigmentoso. Síndrome de Sanctis Cacchione. Presentación de 1 caso. Revista Cubana de Pediatría. [Internet] 2000 [citado el 28 de Noviembre 2017]; 70 (2): 113 - 116. Disponible aquí
9.- Valdovinos B, Sassari M, Scappini M. Xeroderma Pigmentosum (XP). Una realidad presente en la provincia de corrientes. Universidad Nacional del Nordeste. Comunicaciones científicas y tecnológicas. [Internet] [Citado el 28 de Noviembre 2017]. Disponible aquí
10.- Rodríguez R, Aguilar A, Puig P, Solis O, Padilla A. Xeroderma Pigmentoso en dos hermanas. Revista Mexicana de Pediatría. [Internet] 2002 [citado el 28 de Noviembre 2017]; 69 (4): 151 -154. Disponible aquí

¡Gracias por leerme!
Todas las fotos son de mi autoría.
Publicaciones relacionadas
Teratoma inmaduro: Tumor de ovario en edad pediátrica
DIABETES MELLITUS | Todo lo que no sabías explicado de forma sencilla


Saludos, gracias por compartir esta importante y por demás valiosa información, te comento estimada @elocuenciadsnuda, que tu artículo, me hizo recordar el padecimiento de mi mamá de una Carcinoma basocelular, a nivel del ala nasal izquierda, que gracias a dios luego de 2 años de dura lucha, pudimos vencer. Este tipo de aparición en efecto se debe a los excesos de las radiaciones solares a la que estamos expuestos. Saludos.
Gracias a ti por tomarte el tiempo de leerme, de verdad me alegra mucho que tu mamá lo haya superado, saludos y bendiciones para ti y para ella.
enjoy the vote
follow me i vote you when you post i will vote you when i post you also vote me im start following you follow me then you also earn me also earn
Eres doctora? interesante no sabia.... un abrazo que pases un feliz dia mi votico por ti =)
Soy Doc me falta el tora jajajaja aún estoy estudiando, gracias por pasarte por aquí. ¡Saludos!
Excelente aporte @elocuenciadsnuda ...felicitaciones. Gracias por tu contribución a la alfabetización científica de la comunidad.
Muchas gracias Tomas, me alegra que sea bien recibido mi trabajo.
Muy buenos tus aportes a esta comunidad, mil éxitos
Muchas gracias Felix me alegra mucho poder compartir en steemit algo de contenido científico.
This post has been voted on by the steemstem curation team and voting trail.
There is more to SteemSTEM than just writing posts, check here for some more tips on being a community member. You can also join our discord here to get to know the rest of the community!
Hi @elocuenciadsnuda!
Your post was upvoted by utopian.io in cooperation with steemstem - supporting knowledge, innovation and technological advancement on the Steem Blockchain.
Contribute to Open Source with utopian.io
Learn how to contribute on our website and join the new open source economy.
Want to chat? Join the Utopian Community on Discord https://discord.gg/h52nFrV
Hola que gusto pasarme por acá, creo que te comencé a seguir desde tus inicios y es increíble lo mucho de has progresado.
Muy buena información y si duda un caso clínico interesante.
La medicina, no es lo mismo estudiarla que vivirla, creo que pronto también estaré compartiendo algo de contenido medico.
Saludos, una abrazo.
Hooola Astrid si ya ha pasado un tiempito, sería genial estaré atenta leyéndote.