GLYCOGEN STORAGE DISEASE, AN INBORN ERROR OF METABOLISM- Classification,type treatment and diagnoses.

INTRODUCTION
Inborn errors of metabolism can be defined as uncommon genetic disorders which are caused by deficiency in specific proteins that helps in the breaking down of parts of food. The most abundant biomolecules in the human biological system are proteins which are present in the form of hormones, enzymes and various chemical messengers. Deficiency of certain proteins (enzymes) can result in grave consequences which manifest as inborn errors of metabolism. The glycogen storage diseases are disorders of carbohydrate metabolism and it will be the focal point of my discussion for today. Other disorders of intermediary metabolism will be discussed subsequently.

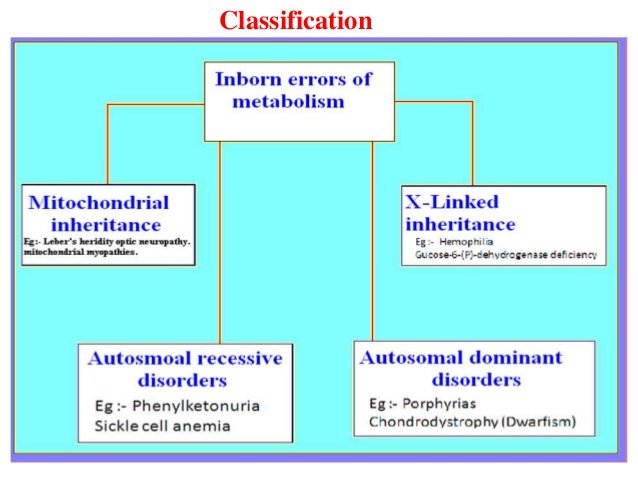
Image B
GENERAL CLASSIFICATION OF INBORN ERRORS OF METABOLISM
The inborn errors as seen in (Image B) above are the generally classified into
Mitochondrial Inheritance, X-linked inheritance, Autosomal recessive disorders and Autosomal dominant disorders.
The inborn errors of metabolism are also classified as disorders of carbohydrate metabolism, amino acid metabolism and organic acid metabolism but hundreds of new inherited metabolic diseases have sufficed and so the classification is more extensive now. Only the major congenital metabolic diseases associated with carbohydrate metabolism will be highlighted however.

{kind=link}
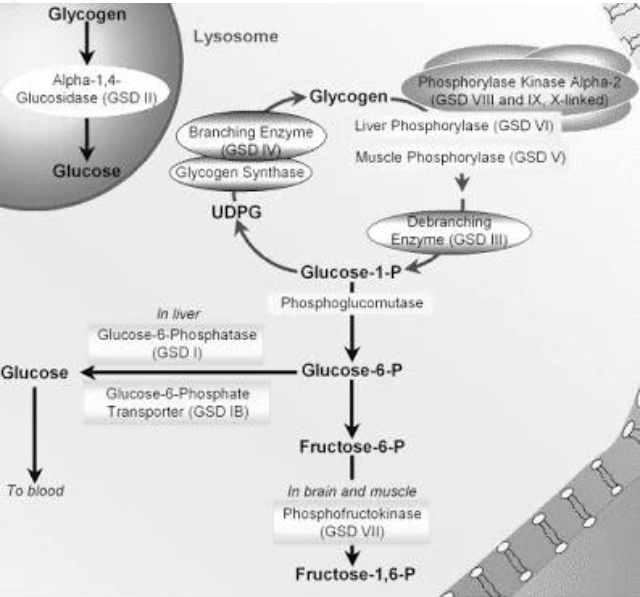
a) GSD 0: this is due to the deficiency of the enzyme Glycogen Synthase. It presents with hyperglycemia, occasional muscle cramping and growth failure in some cases.
b) GSD 1 (Von Gierke’s Disease):this results due to a deficiency in the enzyme glucose-6-phosphatase. Incidence is 1 in 50,000. It presents with hypoglycemia, hyperlipidemia and hepatomegaly. Growth failure, lactic acidosis and hyperuricemia are also other symptoms that are seen in this case.
c) GSD 2 (Pompe’s Disease): this results due to a deficiency in the enzyme acid alpha glucosidase. Incidence is 1 in 40,000 - 50,000. It presents as hepatomegaly. Muscle weakness and heart failure are the major symptoms of Pompe’s Disease.
d) GSD 3 (Cori’s or Forbes’ Disease): this results due to the deficiency of the Glycogen debranching enzyme. Incidence is 1 in 100,000. It presents as hepatomegaly, hyperlipidemia and hypoglycemia. Myopathy is a symptom here.
e) GSD 4 (Andersen Disease):Deficient enzyme is Glycogen branching enzyme. Incidence is 1 in 500,000. It usually presents as hepatomegaly in conjunction with cirrhosis. Failure to thrive is a symptom.
f) GSD 5 (McArdle Disease): the deficient enzyme is muscle Glycogen phosphorylase. Incidence is 1 in 100,000 - 500,000. cramps, rhabdomyolysis and renal failure are the major symptoms.
g) GSD 6 (Her’s Disease): the deficient enzymes are Glycogen phosphorylase and phosphoglycerate mutase. Incidence is 1 in 65,000 - 85,000. This usually presents as hepatomegaly, hypoglycemia and hyperlipidemia. Growth retardation occurs.
h) GSD 7 (Tarui’s Disease): deficient enzyme is muscle phosphofructokinase. Incidence is 1 in 100,000. muscle cramps, weakness causes as a result of exercise and growth retardation are the major symptoms. Haemolytic anemia may also occur.
i) GSD 9:deficient enzyme is phosphorylase kinase. This presents as hepatomegaly, hyperlipidemia and hypoglycemia. Delayed motor development and growth retardation are major symptoms.
j) GSD 10 and GSD 11:deficient enzymes are enolase 3 and muscle lactate dehydrogenase respectively. No symptoms are seen here.
k) GSD XI (Fanconi-Bickel syndrome):This is no longer considered a GSD. The deficient protein here is glucose transporter (GLUT 2). Hypoglycemia and hepatomegaly are seen here.
l) GSD 12 (Aldolase A deficiency):The deficient enzyme here is Aldolase A. Exercise intolerance, cramps, haemolytic anemia and rhabdomyolysis are the symptoms here.
m) GSD 13:deficient enzyme is beta enolase. Exercise intolerance, cramps myalgia are the major symptoms here.
n) GSD 15: deficient enzyme is Glycogenin-1. Muscle atrophy and slowly progressive weakness are the major symptoms.

DIAGNOSIS
Liver biopsy and Hematoxylin and Eosin (H&E) staining are commonly used to detect GSDs.
TREATMENT
Treatment of GSDs is dependent on the type of glycogen storage disease. Consumption of frequent small meals of carbohydrates and cornstarch can help prevent low blood sugar in GSD 1 for example.
Allopurinol and human granulocyte colony stimulating factor may also be used for treatment.
CONCLUSION
The GSDs are very prevalent and come with serious symptoms. They can be acquired or genetic and it has been proven that their manifestations could be triggered by predisposition factors such as lifestyle and eating habits. Thus it is important for us to watch what we eat and what we do as well as exercise regularly to avoid these disorders of metabolism.
THANKS FOR READING !
REFERENCES
The Glycogen Storage Diseases (GSD)
IMAGE SOURCES
THANKS FOR READING !
Insightful... Thanks for sharing this. If I get your point right, that means GSDs are naturally acquired at birth, a genetic thing perhaps.
I'm inclined to ask, though: how fatal is GSD?
There is no medicine as strong as good living and getting good rest. If you obey this rules religiously you have 50% chance of evading sickness and disease. Nice post
very good
Nice. Biochemistry made easy.