Ever Wonder How Proteins Get to the Right Place in Cells?
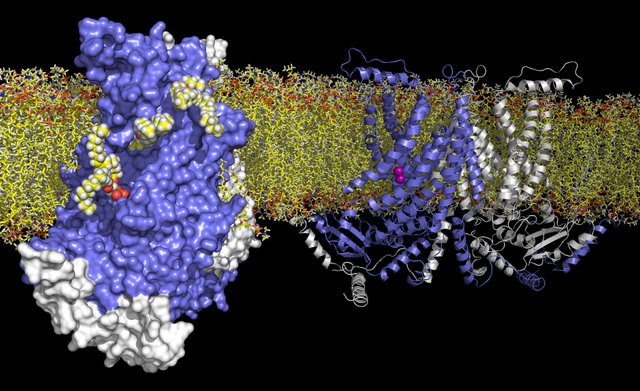
Simply producing the right proteins at the right time is only the first step to regulating cell function on the molecular level.
Equally important is the problem of how proteins find themselves in the right places inside or outside a cell. The post briefly reviews cell structures associated with protein synthesis, including ribosomes, the endoplasmic reticulum (ER), and the Golgi apparatus. It then describes the signal hypothesis proposed by Günter Blobel, which suggests that the initial few amino acids of a protein being synthesized determine whether that protein will be produced in the cytoplasm or in the ER. Concluding by describing other kinds of address labels used to sort proteins and by describing how inclusion-cell disease is caused by malfunctioning protein-sorting mechanisms.

We have seen how the particular suite of proteins found in a cell can be determined by turning on or off the transcription of genes coding for those proteins.
Normal cell function, however, depends not only on having the proper set of proteins, but also on having those proteins occurring in proper locations in the cell. For example, the proteins involved in DNA replication and transcription must find their way into the nucleus of a eukaryotic cell, other proteins must find themselves in the cytoplasm on the cell, others are embedded in the cell’s membranes, and yet others must be exported outside the cell. Thus, another issue that must be considered in order to understand how cells specialize is how proteins become localized in the cell after they are synthesized.
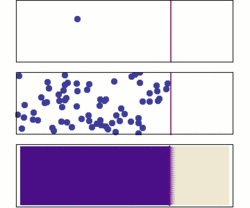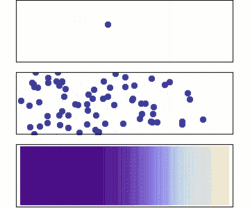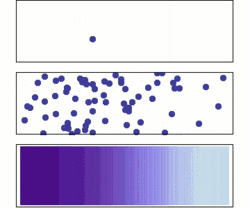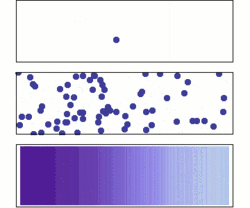
One possible way for proteins to localize would be random diffusion; however, there are two major problems with this idea.
Even in a space as small as a cell, random diffusion would not be very efficient. Some proteins might end up in the right places, but it would take a long time to happen. An even more serious problem is that many of the places proteins need to go are within organelles or subunits of organelles and, thus, are surrounded by biological membranes. Most molecules, especially such molecules as proteins, cannot cross membranes by themselves. To overcome these problems, cells have evolved a variety of protein-trafficking mechanisms.
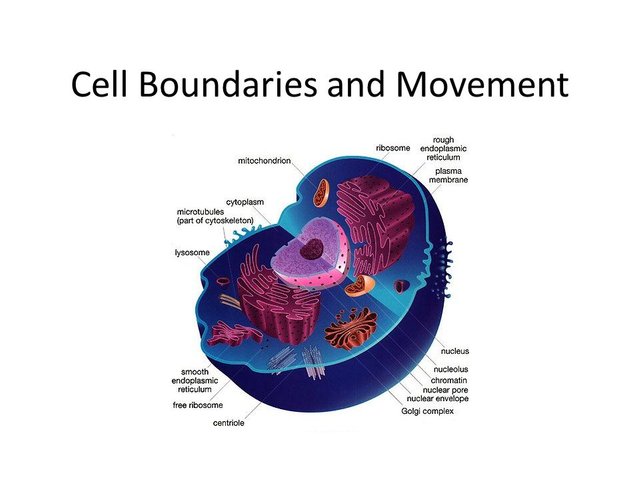
Eukaryotic cells are characterized by having many internal compartments, called organelles, that allow the cell to separate different substances and processes.
One set of organelles is called the endomembrane system, and it has several components. The series of flattened chambers that spreads throughout a cell is called the endoplasmic reticulum (ER), which is interconnected inside. Toward the edges of the cell, these chambers appear to break apart, which is actually the budding off of a number of vesicles from the ER. B. Beyond the ER is a stack of separated, pancake-like chambers, called the Golgi apparatus, which both receives vesicles from the ER and buds off its own vesicles. The overall picture is of two sets of membrane-enclosed chambers with small transport vehicles moving between them and moving outward from the Golgi apparatus. The ER also has bumpy areas as seen under the microscope, called rough ER, which are rough because many ribosomes are attached to the ER surfaces. Ribosomes are large complexes of RNA and protein that construct other proteins. Ribosomes may also be found free in the cytoplasm of the cell. The microscopic anatomy of the endomembrane system has long suggested that at least one of its functions is to process, package, and transport proteins once they have been synthesized. In the 1950s, experiments in which radioactive amino acids were added to a cell in short pulses, then followed over time, confirmed the hypothesis that proteins move from the rough ER to the smooth ER and then to the Golgi apparatus, where they are packaged for transport to the outside of the cell and other places.
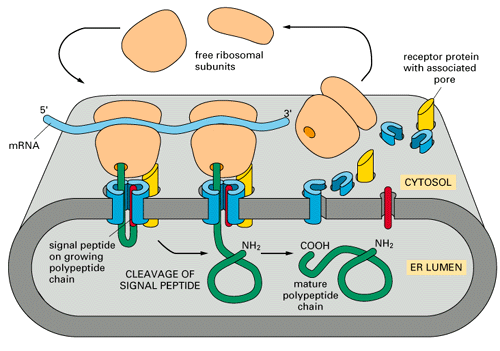
The role of the endomembrane system in processing and transporting proteins raises the question of how proteins are directed into the ER.
In the early 1970s, Günter Blobel proposed the signal hypothesis, which suggests that a sequence of amino acids at the beginning of a polypeptide serves as an address tag that directs a synthesizing ribosome to move to the ER. It is possible to synthesize proteins, even proteins normally secreted into the ER, in a cell-free environment. However, proteins synthesized this way are about two dozen amino acids longer than the same protein synthesized by an intact cell, consistent with the signal hypothesis. Blobel suggested that these extra amino acids are normally removed by the ER because their only function is to direct ribosomes to the ER. This idea was confirmed by adding intact ER to a cell-free solution, which resulted in proteins of the same length as normally synthesized by the cell.
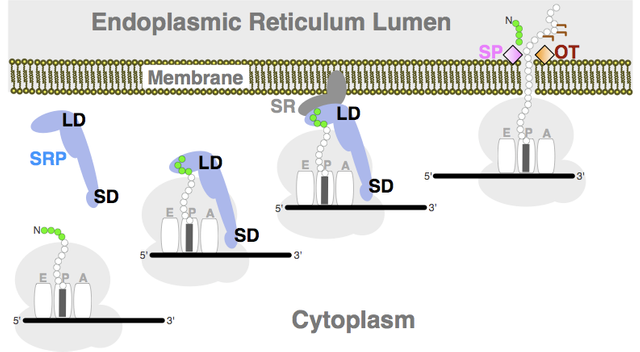
Many experiments since have supported Blobel’s hypothesis and have shown how the address tag works.
All protein synthesis begins in the cytoplasm with free ribosomes. If an address tag occurs at the beginning of the polypeptide, the ribosome stops synthesis. The address tag then attaches to a signal- recognition particle, which docks the ribosome into the ER membrane. Synthesis then restarts, with the synthesized protein directed inside the ER. The information in the address tag ultimately resides in the DNA itself, though these amino acids have nothing to do with the protein’s function.
Proteins synthesized in the ER have many destinations, and similar kinds of address-tag mechanisms are responsible for this additional sorting.
Proteins move progressively through the ER and into the Golgi apparatus via vesicles. In the Golgi apparatus, different kinds of carbohydrates (sugars) are attached to proteins (glycosylation) to form glycoproteins (literally “sugar proteins”). The added sugar components can aid protein function or folding; they also often act as an address tag that determines the protein’s ultimate destination.

Proteins destined for lysosomes are an example of this kind of addressing.
Lysosomes are organelles filled with digestive enzymes that break large molecules into smaller pieces so that other organelles can process them. These digestive enzymes are transported from the Golgi apparatus in vesicles.
Sugars attached to enzymes destined to be transported to lysosomes bind selectively to proteins on the membrane of the Golgi apparatus. These proteins, in turn, bind selectively to other proteins found on the lysosome membrane, so vesicles from the Golgi apparatus will selectively bind to lysosomes if they have lysosomic enzymes inside.
This two-part addressing system, in which proteins have labels that attach to vesicles which themselves have labels, ensures that proteins normally find their proper destinations.
Inclusion-cell (I-cell) disease is an example of a disease caused by dysfunctional protein addressing.
It occurs in about 1 out of every 650,000 births. I-cell disease causes skeletal abnormalities, severe mental retardation, and usually death by age 10. In I-cell disease, lysosomes lack a critical enzyme that allows them to break down waste products. The lysosomes fill up and ultimately destroy the cells containing them. I-cell disease is caused by a single recessive mutation, which suggests that it is caused by a dysfunctional enzyme. Another logical guess would be that the mutation has disabled a crucial transcription factor. However, I-cell patients manufacture the enzyme correctly; it simply never leaves the Golgi apparatus or is secreted outside the cell. Instead, I-cell disease is caused by the lack of an enzyme responsible for attaching a specific carbohydrate address tag. This missing address tag prevents the digestive enzyme from becoming packaged in a vesicle that will deliver it to the lysosome.

BTC: 1GfXFX1o1uUkC4HuGuKUPty6wdu6oi8X1t 


I am a follower of his notes, his work is very good, congratulations on another material magnificent thank you very much for sharing