Plant Genomic DNA Extraction using CTAB
Introduction
The search for a more efficient means of extracting DNA of both higher quality and yield
has lead to the development of a variety of protocols, however the fundamentals of DNA
extraction remains the same. DNA must be purified from cellular material in a manner
that prevents degradation. Because of this, even crude extraction procedures can still be
adopted to prepare a sufficient amount of DNA to allow for multiple end uses.
DNA extraction from plant tissue can vary depending on the material used. Essentially
any mechanical means of breaking down the cell wall and membranes to allow access to
nuclear material, without its degradation is required. For this, usually an initial grinding
stage with liquid nitrogen is employed to break down cell wall material and allow access
to DNA while harmful cellular enzymes and chemicals remain inactivated. Once the
tissue has been sufficiently ground, it can then be resuspended in a suitable buffer, such
as CTAB. In order to purify DNA, insoluble particulates are removed through
centrifugation while soluble proteins and other material are separated through mixing
with chloroform and centrifugation. DNA must then be precipitated from the aqueous
phase and washed thoroughly to remove contaminating salts. The purified DNA is then
resuspended and stored in TE buffer or sterile distilled water. This method has been
shown to give intact genomic DNA from plant tissue. To check the quality of the
extracted DNA, a sample is run on an agarose gel, stained with ethidium bromide, and
visualised under UV light.
Materials
CTAB buffer
Microfuge tubes
Mortar and Pestle
Liquid Nitrogen
Microfuge
Absolute Ethanol (ice cold)
70 % Ethanol (ice cold)
7.5 M Ammonium Acetate
55o C water bath
Chloroform : Iso Amyl Alcohol (24:1)
Water (sterile)
Agarose
6x Loading Buffer
1x TBE solution
Agarose gel electrophoresis system
Ethidium Bromide solution
CTAB buffer 100ml
2.0 g CTAB (Hexadecyl trimethyl-ammonium bromide)
10.0 ml 1 M Tris pH 8.0
4.0 ml 0.5 M EDTA pH 8.0 (EthylenediaminetetraAcetic acid Di-sodium salt)
28.0 ml 5 M NaCl
40.0 ml H2O
1 g PVP 40 (polyvinyl pyrrolidone (vinylpyrrolidine homopolymer) Mw 40,000)
Adjust all to pH 5.0 with HCL and make up to 100 ml with H2O.
1 M Tris pH 8.0
Dissolve 121.1 g of Tris base in 800 ml of H2O. Adjust pH to 8.0 by adding 42 ml of
concentrated HCL. Allow the solution to cool to room temperature before making the
final adjustments to the pH. Adjust the volume to 1 L with H2O. Sterilize using an
autoclave.
5x TBE buffer
54 g Tris base
27.5 g boric acid
20 ml of 0.5M EDTA (pH 8.0)
Make up to 1L with water.
To make a 0.5x working solution, do a 1:10 dilution of the concentrated stock.
1% Agarose gel
1 g Agarose dissolved in 100 ml TBE
Procedure
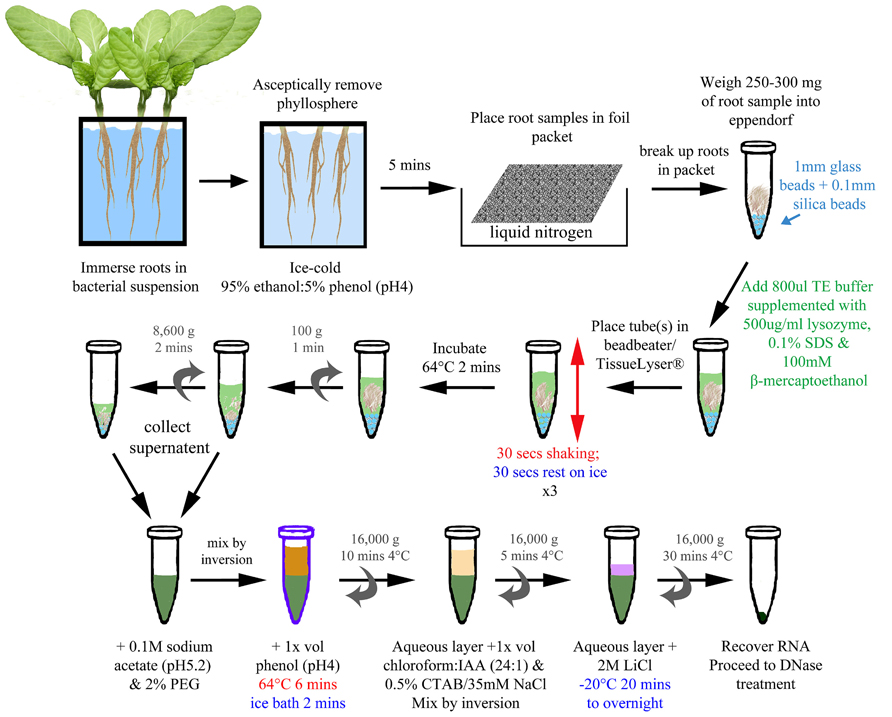
- Grind 200 mg of plant tissue to a fine paste in approximately 500 μl of CTAB buffer.
- Transfer CTAB/plant extract mixture to a microfuge tube.
- Incubate the CTAB/plant extract mixture for about 15 min at 55o C in a recirculating
water bath.
- After incubation, spin the CTAB/plant extract mixture at 12000 g for 5 min to spin
down cell debris. Transfer the supernatant to clean microfuge tubes.
- To each tube add 250 μl of Chloroform : Iso Amyl Alcohol (24:1) and mix the
solution by inversion. After mixing, spin the tubes at 13000 rpm for 1 min.
- Transfer the upper aqueous phase only (contains the DNA) to a clean microfuge
tube.
- To each tube add 50 μl of 7.5 M Ammonium Acetate followed by 500 μl of ice cold
absolute ethanol.
- Invert the tubes slowly several times to precipitate the DNA. Generally the DNA can
be seen to precipitate out of solution. Alternatively the tubes can be placed for 1 hr at
-20 o C after the addition of ethanol to precipitate the DNA.
- Following precipitation, the DNA can be pipetted off by slowly rotating/spinning a
tip in the cold solution. The precipitated DNA sticks to the pipette and is visible as a
clear thick precipitate. To wash the DNA, transfer the precipitate into a microfuge
tube containing 500 μl of ice cold 70 % ethanol and slowly invert the tube. Repeat.
((alternatively the precipitate can be isolated by spinning the tube at 13000 rpm for a
minute to form a pellet. Remove the supernatant and wash the DNA pellet by adding
two changes of ice cold 70 % ethanol )).
- After the wash, spin the DNA into a pellet by centrifuging at 13000 rpm for 1 min.
Remove all the supernatant and allow the DNA pellet to dry (approximately 15 min).
Do not allow the DNA to over dry or it will be hard to re-dissolve.
- Resuspend the DNA in sterile DNase free water (approximately 50-400 μl H2O; the
amount of water needed to dissolve the DNA can vary, depending on how much is
isolated). RNaseA (10 μg/ml) can be added to the water prior to dissolving the DNA
to remove any RNA in the preparation (10 μl RNaseA in 10ml H2O).
- After resuspension, the DNA is incubated at 65o C for 20 min to destroy any DNases
that may be present and store at 4o C.
- Agarose gel electrophoresis of the DNA will show the integrity of the DNA, while
spectrophotometry will give an indication of the concentration and cleanliness.
DNA quality confirmation
– Prepare a 1 % solution of agarose by melting 1 g of agarose in 100 mL of 0.5x TBE
buffer in a microwave for approximately 2 min. Allow to cool for a couple of
minutes then add 2.5 μl of ethidium bromide, stir to mix.
– Cast a gel using a supplied tray and comb. Allow the gel to set for a minimum of 20
min at room temperature on a flat surface.
– Load the following into separate wells
o 10 μL 1kb ladder
o 5 μL sample + 5 μL water + 2 μL 6x Loading Buffer
– Run the gel for 30 min at 100 V
– Expose the gel to UV light and photograph (demonstration)
– Confirm DNA quality, presence of a highly resolved high molecular weight band
indicates good quality DNA, presence of a smeared band indicates DNA
degredation.
Congratulations @genetics.life! You received a personal award!
Click here to view your Board
Congratulations @genetics.life! You received a personal award!
You can view your badges on your Steem Board and compare to others on the Steem Ranking
Vote for @Steemitboard as a witness to get one more award and increased upvotes!