"Cystic fibrosis": clinical history real
This is one of the cases that draws much attention since it is not very frequent that this type of patient comes to the emergency of adults, and this is because the survival rate of these patients is up to a maximum of 10 years in underdeveloped countries; where years ago their expectations of lives were very low to only 6 months of born due to the multiple complications that originate the disease. Survival has now risen, reaching a maximum of 48 years. Now I tell you in this opportunity:
It is a 16-year-old female patient with a history of cystic fibrosis diagnosed at 5 years of age, who started the current disease approximately two weeks ago and is characterized by progressive lower limb volume and weakness generalized, it would also be associated with the clinical picture; two days prior productive cough abundant purulent and fetid concomitant chills, reason for which comes to the emergence of adults, is valued and is entered.
At the physical examination of the patient in normal clinical conditions afebrile to the accentuated dehydrata dehydrata touch, mucosal cutaneous paleness, symmetrical thorax, hyperexpansible MVS audible in AsCsPs with bilateral crackles and wheezing, as well as grade IV / IV edema in lower limbs with paraclinica that reported leucositosis a expense of segmented with elevated hepatic transaminases (tGo and tgp) bilirubins, hyperglycemia ranging from 150 to 180 fasting with a marked hypoalbumine.
Treatment with broad-spectrum antibiotic therapy, respiratory therapies, control paraclinics, uroanalysis, ph, and gasarterial therapy, abodomino-pelvic echo and gastroenterology assessment are initiated. In view of the fact that this pathology is not frequent in young adults, this is due to the aforementioned who values it in a gastro-pediatrician and in view of the clinic of the patient makes reference to it is very probable that its pathology of base cystic fibrosis pulmonary level, in case this young man has begun to take other organs as white as liver and pancreas, and that he needs to perform a series of tests and start treatment for his pathology. Once stabilized and compensated by the respiratory part, and with diminished edema that presented in lower limbs, it moves to floor or room of hospital attention to continue the studies; nowadays the young girl is in the said room with medical treatment.

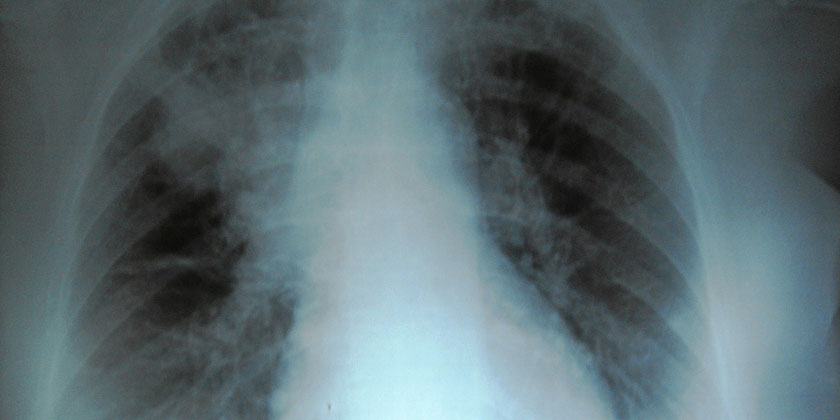
Photo taken from the hospital where I work, in precarious conditions.
Now I would like to explain the theoretical part of this very rare rare disease as I already mentioned in the medical record; in order that you understand a little better that this type of disease is treated.
Cystic fibrosis is an inherited disease of the mucous and sweat glands. It mainly affects the lungs, pancreas, liver, intestines, sinuses and sexual organs. Cystic fibrosis makes the mucus thick and sticky. This mucus covers the lungs, causing problems to breathe and facilitating the growth of bacteria. This can lead to repeated lung infections and lung damage.
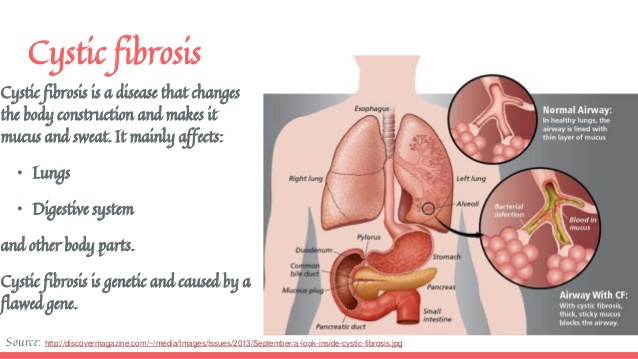
https://www.slideshare.net/AlexandraBejanyan/cystic-fibrosis-brief-explanation
Symptom
http://www.webconsultas.com/salud-al-dia/fibrosis-quistica/sintomas-de-la-fibrosis-quistica
There are indicative signs that, at an early age, may induce us to think that we are facing an affection of cystic fibrosis, according to Giron. These signs can be:
Salty sweat: by affecting the sweat glands. "This can lead to hyponatraemic and hypochloremic dehydration in hot weather," he says.
Pulmonary symptoms: Consistent of cough with expectoration, with frequent respiratory infections that deteriorate the respiratory capacity.
Nasal symptoms: With rhinitis, sinusitis and nasal polyposis.
Digestive symptoms in a high percentage of patients: "This occurs with the presence of pancreatic insufficiency and malabsorption of fats, leading to a deterioration of nutritional status," says Giron. "With the progression of the pancreatic affectation, diabetes originates, which complicates the evolution of the disease".
Infertility in men due to obstructive azoospasm and decreased fertility in women.
Types
There are several classifications of cystic fibrosis:
According to the pulmonary gravity is classified as mild, moderate and severe.
Pancreatic involvement is classified as pancreatic insufficiency and insufficiency.
According to the mutations. Depending on the alteration that originate in the CFTR protein, they are classified into 6 classes. This classification is currently important given the development of therapies directed to the CFTR protein, mainly enhancers and correctors.
Causes
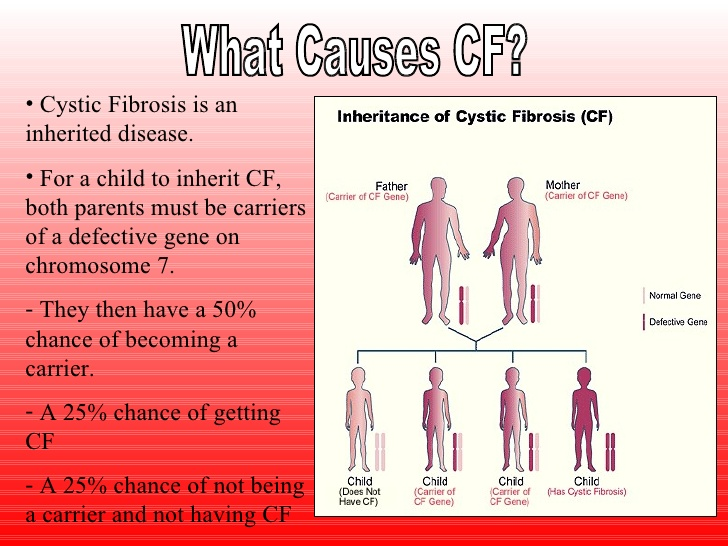
As Girón explains, the cystic fibrosis transmembrane conductance regulator gene (CFTR), under normal conditions, encodes the CFTR protein of the chloride epithelial channel, responsible for regulating the absorption and secretion of salt and water in several organic systems, including the lungs, pancreas, intestinal tract, biliary tract, sweat glands and reproductive tract.
"In cystic fibrosis, the lack of regulation of the transport of chloride in these organs results in the multisystemic pathology associated with this disease," explains the pneumologist at Separ. "Therefore, cystic fibrosis is a systemic disorder, not just a pulmonary pathology."
https://www.slideshare.net/jenny1tafe/cystic-fibrosis-1945416
Treatment
-Malnutrition and digestive system problems:
People with cystic fibrosis absorb the fats poorly, resulting in an important state of malnutrition, which must be countered by a high-calorie diet with a fat content that is somewhat higher than normal (120-150% of the calories normally recommended), and provide enough proteins and fat-soluble vitamins (A and E) ..
In order to control the nutritional status and to act according to the needs of the patient, biochemical determinations (blood count, proteins, fats ...), and weight, height, arm perimeter must be carried out periodically ...
Patients with pancreatic insufficiency should also consume commercial pancreatic enzyme preparations.
The treatment of meconium ileus consists of surgery.
-Respiratory system:
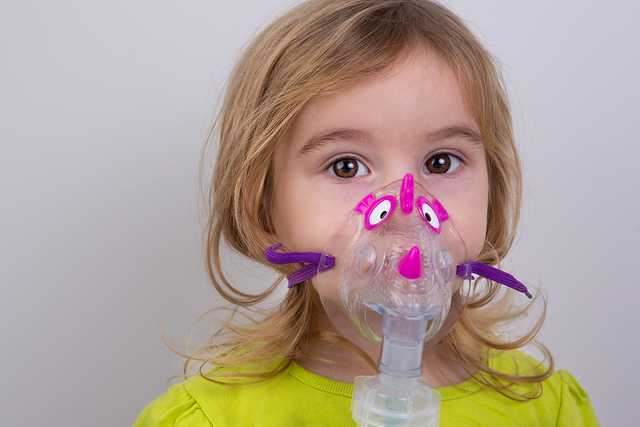
The purpose of treatment of respiratory problems is to return secretions more fluid, to avoid bronchial obstruction, and to prevent and treat infections. In addition, respiratory physiotherapy is recommended, with postural measures that facilitate the expulsion of secretions (2-3 daily sessions of 20 minutes before meals) and intermittent administration of drugs such as aerosolized bronchodilators (the same ones used by asthmatics) and mucolytics (to facilitate removal of secretions).
In order to treat respiratory infection, antibiotics are administered mainly to the most involved microorganisms such as Staphylococcus aureus and Pseudomona aeruginosa. In most cases, they establish themselves as habitual colonizers of the airway, being extremely difficult to eradicate them.
If the infections are persistent, the antibiotic treatment must be continued, but if they are infrequent the treatment will be limited to the time necessary in each case. Pulmonary transplantation may be indicated in patients with highly evolved disease.
It is necessary that the patient ingests salt supplements to cope with sweat losses, especially during hot weather.
Given the chronic and incurable nature of the disease, it is advisable for patients and their families to receive psychological support.
Multiple therapies targeting the genetic mutation that causes the disease are being tested, with no definitive results yet.
http://www.sigmalive.com/en/lifestyle/health/129778/quotgroundbreakingquot-cystic-fibrosis-treatment
Diagnosis
Early diagnosis is essential to assign appropriate treatment as soon as possible.
http://www.cuidateplus.com/enfermedades/respiratorias/fibrosis-quistica.html
The direct relationship between an early diagnosis and the development of a better quality of life in the patients is demonstrated. The ideal system for the detection of the pathology is the realization of a neonatal screening, a simple blood test that can indicate the possibility of having cystic fibrosis. Unfortunately, this system is not implemented by all health administrations, mainly because of the costs and the possibility of false positives.
"Prior to neonatal screening, patients were diagnosed for clinical symptoms: positive sweat test (> 60mEq / l) and identification of mutations compatible with cystic fibrosis. At present, with neonatal screening and a very early diagnosis the clinic is not necessary, "says Girón.
The specialist points out that, once the diagnosis is established, it is important to carry out tests for respiratory monitoring (microbiological samples of the respiratory tract, pulmonary function studies and imaging studies), digestive monitoring (intestinal absorption tests), screening diabetes, bone and nutritional disorders.
Prevention
Cystic fibrosis is a genetic disease, that is, people are born with it. For this reason it can not be prevented, unless it is known that the parents are carriers and a selection of the embryo is made in cases in which the patients wish to have offspring.
"As for the prevention of symptoms, with the current treatments slows down a little the lung," says Giron. "There are currently some approved treatments in a type of mutation that call mutations 'opening the channel' before which there is a specific treatment that seems to prevent the symptoms."
Forecast
The life expectancy of patients with cystic fibrosis has improved greatly in recent years, since it has gone from four years in 1950 to 25 years in 1990. And it is currently around 39 years. This is mainly due to early diagnosis, improvements in maintenance of nutritional status and advances in the treatment of respiratory infections.
However, despite the progress made, the most frequent cause of death in patients with cystic fibrosis is usually associated with malnutrition caused by malabsorption of fats and nutrients, due to exocrine pancreatic insufficiency, recurrence of respiratory infections and lack of appetite.
Other less frequent causes of death are meconium ileus, chronic respiratory failure, heart failure, and impaired hepatic function.
Progress in transplantation has also improved the life expectancy of these people, not only in terms of increased donations, but also improved surgical techniques and the development of more effective drugs for post-transplant immunosuppression.
Credits:
https://medlineplus.gov/spanish/cysticfibrosis.html
http://www.webconsultas.com/fibrosis-quistica/pronostico-de-la-fibrosis-quistica-760
http://www.cuidateplus.com/enfermedades/respiratorias/fibrosis-quistica.html
Remember the health is the most important, for more people are very busy with their work, it is advisable to always visit your doctor and get checked periodically, if you want to have a healthy life do it.
And remember that the best medicine is God, without the doctors we could not save lives.
Greetings from Venezuela.
By: Johana Albarran
Doctor
I'm happy to come by and upvote my favorite blogger Joha!! ;-D
jejej I'm really your favorite?
:-D happy to say yes
you know that he is also my favorite, you were the first person to support me when I arrived at steemit and that is very pleasing and the best of all is that we form a friendship and hope it lasts a long time
A very long time indeed!! :-D