POMPE DISEASE
Pompe disease is an inherited disorder caused by the buildup of complex sugar called glycogen in the body's cells. Pompe Disease also known as acid alpha glucosidase (GAA) deficiency, acid maltase deficiency (AMD), glycogen storage disease type II.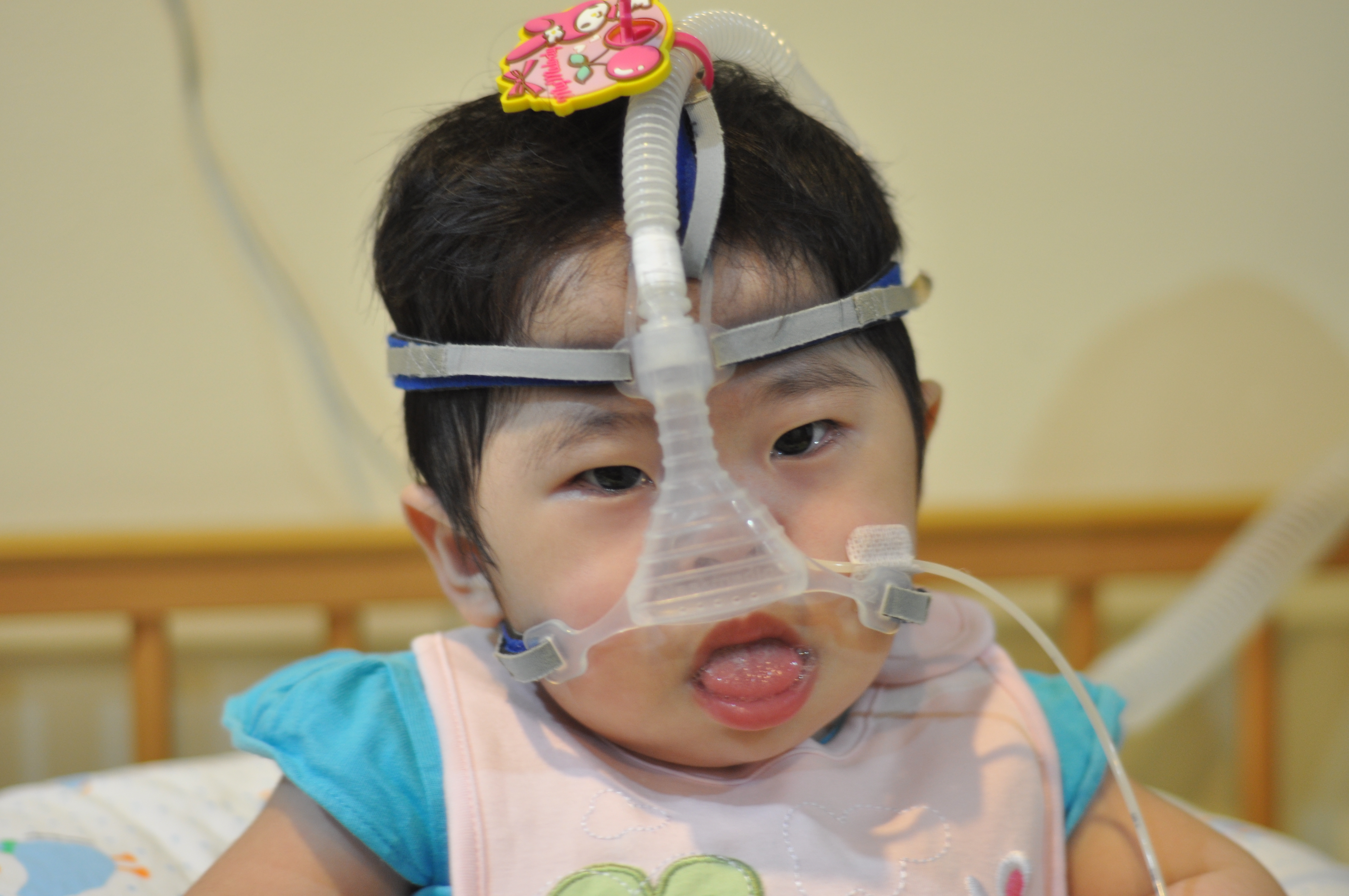
image source: http://www.rdss.org.sg/wp-content/uploads/2014/04/13
The accumulation of glycogen in certain organs and tissues, especially muscles, impairs their ability to function normally. The disease is named after Johannes C. Pompe, a Dutch doctor who first described the disorder in 1932 in an infant patient. Pompe can affect people of all ages, with symptoms first occurring at any time from infancy to adulthood. Pompe disease causes muscle weakness and trouble breathing. It mostly affects the liver, heart, and muscles.
image source: http://cdn.smdailyjournal.com/article_image/smdj_article_123584_1
Cause
Pompe disease is inherent from parents to children. To get it, two flawed genes are inherited, one from each parent.
The disease is caused by a mutation of the acid alpha-glucosidase gene (GAA) gene. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%.
image source: http://www.brianjorlowski.com/Samples/Illustrator/Slide%205%20Fig%20pt2
Symptoms
- Trouble eating and not gaining weight
- Poor head and neck control
- Rolling over and sitting up later than expected
- Breathing problems and lung infections
- Enlarged and thickening heart or heart defects
- Enlarged liver
- Enlarged tongue
- Most patients experience muscle symptoms, such as weakness and cramps, although certain glycogen storage diseases manifest as specific syndromes, such as hypoglycemic seizures or cardiomegaly.

image source: http://tommythedoc.weebly.com/uploads/1/4/0/4/14044765/380650442_orig

Pathophysiology
With an enzyme defect, carbohydrate metabolic pathways are blocked, and excess glycogen accumulates in affected tissues. Each GSD represents a specific enzyme defect, and each enzyme is either in specific sites or is in most body tissues.
Acid maltase is a lysosomal enzyme that catalyzes the hydrogenation of branched glycogen compounds, notably maltose, to glucose. The conversion generally is a one-way reaction from glycogen to glucose-6-phosphate. When acid maltase is deficient, glycogen accumulates within tissues. Acid maltase is found in all tissues, including skeletal and cardiac muscle. Accumulation of glycogen in cardiac muscle leads to cardiac failure in the infantile form.
Diagnosis
A diagnosis of Pompe disease is based upon a thorough clinical evaluation, a detailed patient and family history, and a variety of tests. Prenatal diagnosis is possible when a pregnancy is believed to be at risk for Pompe disease. Also it depends on muscle biopsy, electromyelography, the ischemic forearm test, creatine kinase levels, patient history, and physical examination findings.
Mortality/Morbidity
The infantile form usually is fatal, with most deaths occurring within 1 year of birth. Cardiomegaly with progressive obstruction to left ventricular outflow is a major cause of mortality. Later clinical onset usually corresponds with more benign symptoms and disease course.
The adult form manifests with dystrophy and respiratory muscle weakness. Respiratory insufficiency is a significant morbidity.
Treatment
Treatment may require the coordinated efforts of a team of specialists with expertise in treating neuromuscular disorders.
Early treatment, is key to holding off the damage in the body.
Two medications replace the missing protein and help your body process sugar correctly.
- Myozyme, for babies and children
- Lumizyme
- No cure exists, although diet therapy and enzyme replacement therapy may be highly effective at reducing clinical manifestations
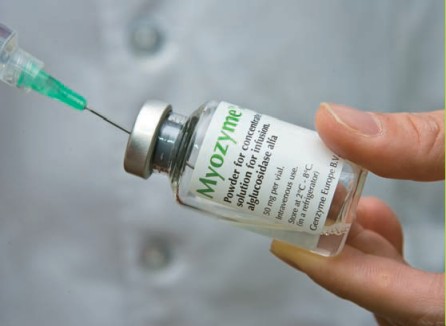
image source: https://userscontent2.emaze.com
References
- van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet. 2008 Oct 11;372(9646):1342-53. doi: 10.1016/S0140-6736(08)61555-X. Review. Citation on PubMed
- https://en.wikipedia.org/wiki/Glycogen_storage_disease_type_II
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, Crowley JF, Downs S, Howell RR, Kravitz RM, Mackey J, Marsden D, Martins AM, Millington DS, Nicolino M, O'Grady G, Patterson MC, Rapoport DM, Slonim A, Spencer CT, Tifft CJ, Watson MS. Pompe disease diagnosis and management guideline. Genet Med. 2006 May;8(5):267-88. Erratum in: Genet Med. 2006 Jun;8(6):382. ACMG Work Group on Management of Pompe Disease [removed]; Case, Laura [corrected to Case, Laura E]. Citation on PubMed or Free article on PubMed Central
- Van Capelle CI, Goedegebure A, Homans NC, et al. Hearing loss in Pompe disease revisited: results from a study of 24 children. J Inherit Metab Dis. 2010;33:597-602. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2946566/