Atomic And Molecular Clusters Latest Preprints | 2019-06-16
Atomic And Molecular Clusters
A Novel Discrete Theory of a Screw Dislocation in the BCC Crystal Lattice (1906.04332v1)
Shigeki Matsutani
2019-06-11
In this paper, we proposed a novel method using the elementary number theory to investigate the discrete nature of the screw dislocations in crystal lattices, simple cubic (SC) lattice and body centered cubic (BCC) lattice, by developing the algebraic description of the dislocations in the previous report (Hamada, Matsutani, Nakagawa, Saeki, Uesaka, Pacific J. Math.~for Industry {\bf{10}} (2018), 3). Using the method, we showed that the stress energy of the screw dislocations in the BCC lattice and the SC lattice are naturally described; the energy of the BCC lattice was expressed by the truncated Epstein-Hurwitz zeta function of the Eisenstein integers, whereas that of SC lattice is associated with the truncated Epstein-Hurwitz zeta function of the Gauss integers.
Generation and Characterization of Tailored MIR Waveforms for Steering Molecular Dynamics (1906.03519v1)
Markus A. Jakob, Mahesh Namboodiri, Mark J. Prandolini, Tim Laarmann
2019-06-08
The dream of physico-chemists to control molecular reactions with light beyond electronic excitations pushes the development of laser pulse shaping capabilities in the mid-infrared (MIR) spectral range. Here, we present a compact optical parametric amplifier platform for the generation and shaping of MIR laser pulses in the wavelength range between
m and
m. Opportunities for judiciously tailoring the electromagnetic waveform are investigated, demonstrating light field control with a spectral resolution of 60 GHz at a total spectral bandwidth of 5 THz. In experiments focusing on spectral amplitude manipulation these parameters result in a time window of 1.8 ps available for shaping the temporal pulse envelope and a phase modulation resolution of 100 mrad for several picosecond delays.


Efimov universality with Coulomb interaction (1901.03643v3)
C. H. Schmickler, H. -W. Hammer, E. Hiyama
2019-01-11
The universal properties of charged particles are modified by the presence of a long-range Coulomb interaction. We investigate the modification of Efimov universality as a function of the Coulomb strength using the Gaussian expansion method. The resonant short-range interaction is described by Gaussian potentials to which a Coulomb potential is added. We calculate binding energies and root mean square radii for the three- and four-body systems of charged particles and present our results in a generalised Efimov plot. We find that universal features can still be discerned for weak Coulomb interaction, but break down for strong Coulomb interaction. The root-mean-square radius plateaus at increasingly smaller values for strong Coulomb interaction and the probablity distributions of the states become more concentrated inside the Coulomb barrier. As an example, we apply our universal model to nuclei with an alpha-cluster substructure. Our results point to strong non-universal contributions in that sector.
The Ratio Law of the Structure Evolution and Stability for Ti O
O (
(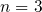 -
-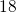 ,
, 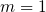 -
-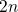 ) Clusters (1906.05354v1)
) Clusters (1906.05354v1)
Hongbo Du, Yu Jia, Chunyao Niu, Kaige Hu, Haifeng Li, Lingmin Yu
2019-06-03
Most theoretical investigations about titanium oxide clusters focus on (TiO
)
are produced experimentally. In this work, first-principles calculations are performed to probe the evolution of Ti
, there exist one relatively stable specie; while for
-








Crystalline droplets with emergent topological color-charge in many-body systems with sign-changing interactions (1905.13217v1)
P. Karpov, F. Piazza
2019-05-30
We introduce a novel type of self-bound droplet which carries an emergent color-charge. We consider a system of particles hopping on a lattice and interacting via a commensurately sign-changing potential which is attractive at short range. The droplet formation is heralded by spontaneous crystallization into topologically distinct domains. This endows each droplet with an emergent color-charge governing their mutual interactions: attractive for equal colors and repulsive otherwise. The number of allowed colors is fixed only by the discrete spatial symmetries of the interaction potential. With increasing interaction range, the droplets become progressively more mobile, with their color-charge still being energetically protected, allowing for non-trivial viscous dynamics of the interacting droplet plasmas formed during cooling. Sign-changing potentials with short-range attraction appear quite naturally for light-mediated interactions and we concretely propose a realization in state-of-the-art experiments with cold atoms in a multimode optical cavity.
Dissociation dynamics in the dissociative electron attachment to ammonia molecule (1905.10993v1)
Dipayan Chakraborty, Aranya Giri, Dhananjay Nandi
2019-05-27
Complete dissociation dynamics of low energy electron attachment to ammonia molecule has been studied using velocity slice imaging (VSI) spectrometer. One low energy resonant peak around 5.5 eV and a broad resonance around 10.5 eV incident electron energy has been observed. The resonant states mainly dissociate via H
and NH
fragments, though for the upper resonant state, signature of NH
symmetry in the 10.5 eV resonance energy whereas, the 5.5 eV resonance is associated with the well known A



The water-carbon monoxide dimer: new infrared spectra, ab initio rovibrational energy level calculations, and an interesting intermolecular mode (1905.10903v1)
A. Barclay, A. van der Avoird, A. R. W. McKellar, N. Moazzen-Ahmadi
2019-05-26
Rovibrational energy level calculations using a high-level intermolecular potential surface are reported for H2O-CO and D2O-CO. They predict the ground K = 1 levels to lie about 20 (12) cm-1 above K = 0 for H2O-CO (D2O-CO) in good agreement with past experiment. But the first excited K = 1 levels are predicted to lie about 3 cm-1 below their K = 0 counterparts in both cases. Intensity calculations also indicate that mid-infrared transitions from the K = 0 ground state to this seemingly anomalous excited K = 1 state should be observable. These predictions are strikingly verified by new spectroscopic measurements covering the C-O stretch region around 2200 cm-1 for H2O-CO, D2O-CO, and HOD-CO, and the O-D stretch region around 2700 cm-1 for D2O-CO, HOD-CO, and DOH-CO. The experiments probe a pulsed supersonic slit jet expansion using tunable infrared quantum cascade laser or optical parametric oscillator sources. Discrete perturbations in the O-D stretch region give an experimental lower limit of about 340 cm-1 for D2O-CO, as compared to our calculated binding energy of 368 cm-1. Wavefunction plots are presented to help understand the intermolecular dynamics of H2O-CO. Coriolis interactions are invoked to explain the seemingly anomalous energies of the first excited K = 1 levels.
Photoionization of Molecular Endohedrals (1905.09739v1)
M. Ya. Amusia, L. V. Chernysheva, S. K. Semenov
2019-05-23
We calculate the photoionization cross-section of a molecular endohedral. We limit ourselves to two-atomic molecules. The consideration is much more complex than for atomic endohedrals because the system even for almost spherical fullerenes has only cylindrical instead of spherical symmetry. On the other hand, molecular endohedral is more interesting since the interelectron interaction in molecules is relatively stronger than in similar atoms. We present here results of calculations of molecular hydrogen stuffed inside almost spherical fullerene. For comparison, we perform calculations also for atomic endohedral with Helium inside fullerene. The results are obtained both in the single-electron Hartree-Fock approximation and with account of multi-electron correlations in the frame of so-called random phase approximation with exchange. The presence of the fullerenes shell results in prominent oscillations in the endohedrals photoionization cross section. The role of interelectron correlations becomes clear by comparing HF and RPAE results for molecular and atomic endohedral on the one side with that for corresponding isolated molecule and atom on the other.
Metrology of time-domain soft X-ray attosecond pulses and re-evaluation of pulse durations of three recent experiments (1905.09526v1)
Zhao Xi, Wang Su-Ju, Yu Wei-Wei, Wei Hui, Lin C. D
2019-05-23
Attosecond pulses in the soft-X-ray (SXR) to water-window energy region offer the tools for creating and studying target specific localized inner-shell electrons or holes in materials, enable monitoring or controlling charge and energy flows in a dynamic system on attosecond timescales. Recently, a number of laboratories have reported generation of continuum harmonics in the hundred-electron-volt to kilovolt region with few-cycle long-wavelength mid-infrared lasers. These harmonics have the bandwidth to support pulses with duration of few- to few-ten attoseconds. But harmonics generated in a gas medium have attochirps that cannot be fully compensated by materials over a broad spectral range; thus, realistically what are the typical shortest attosecond pulses that one can generate? To answer this question, it is essential that the temporal attosecond pulses be accurately characterized. By re-analyzing the soft X-ray harmonics reported in three recent experiments \cite{chang_natcom2017,Thomas_OE2017,Bieger_2017PRX} using a newly developed broadband phase retrieval algorithm, we show that their generated attosecond pulses are all longer than about 60 as. Since broadband pulses tend to have high-order chirps away from the spectral center of the pulse, the algorithm has to be able to retrieve accurately the phase over the whole bandwidth. Our re-evaluated pulse durations are found to be longer than those previously reported. We also introduce the autocorrelation (AC) of the streaking spectrogram. By comparing the ACs from the experiments and from the retrieved SXR pulses, the accuracy of the retrieved results can be directly visualized to ensure that correct phases have been obtained. Our retrieval method is fast and accurate, and it shall provide a powerful tool for the metrology of few-ten-attosecond pulses in the future.
First Principles Study of Structural and Optical Properties of B Isomers (1802.01072v2)
Isomers (1802.01072v2)
Pritam Bhattacharyya, Ihsan Boustani, Alok Shukla
2018-02-04
In this work we undertake a comprehensive numerical study of the ground state structures and optical absorption spectra of isomers of B
symmetry. In bulk boron icosahedron is the basic structural unit, but, our vibrational frequency analysis reveals that it is unstable in the isolated form. We speculate that this instability could be due to Jahn-Teller distortion because five-fold degenerate HOMO orbitals in


flagged for bid bot abuse @steemflagrewards
Steem Flag Rewards mention comment has been approved! Thank you for reporting this abuse, @mathowl.
You bought votes to increase the rewards of your post above the value of its content.
This post was submitted via our Discord Community channel. Check us out on the following link!
SFR Discord
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.
Follow on flag for bid bot abuse @steemflagrewards.